Knowing Your NGS Upstream: Alignment and Variants
Date: March 27, 2013
Presenter: Gabe Rudy, Vice President of Product Development
Duration: 120 minutes
ACCESS THIS WEBCAST :


Abstract
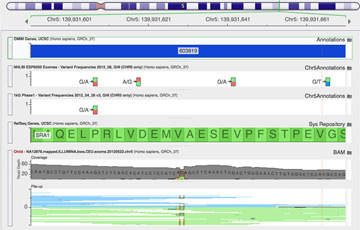
Alignment algorithms are not just about placing reads in best-matching locations to a reference genome. They are now being expected to handle small insertions, deletions, gapped alignment of reads across intron boundaries and even span breakpoints of structural variations, fusions and copy number changes. At the same time, variant-calling algorithms can only reach their full potential by being intimately matched to the aligner's output or by doing local assemblies themselves. Knowing when these tools can be expected to perform well and when they will produce technical artifacts or be incapable of detecting features is critical when interpreting any analysis based on their output.
This presentation will compare the performance of the alignment and variant calling tools used by sequencing service providers including Illumina Genome Network, Complete Genomics and The Broad Institute. Using public samples analyzed by each pipeline, we will look at the level of concordance and dive into investigating problematic variants and regions of the genome.