In the 1990s the genetic industry voiced a request for a variant catalog that incorporates associated variant information such as phenotypic and metabolic pathways. The call was answered by NCBI, which created dbSNP; dbSNP became publicly available in 1998 and around 1.5 million variants. Fast forward to the present and dbSNP now contains over 2 billion SNPs spanning human, rat,… Read more »
We have now reached the final blog of the NGS-Solutions for Clinical Variant Analysis series. Part I of this series explored the capture of variant classifications in the VSClinical environment when following the ACMG and AMP guidelines. Part II was similar in content but for the capture of clinically relevant copy number variants as well as using a CNV catalog… Read more »
The potential of genetic testing to impact a patient’s life has been greatly accelerated by the sharing of variant interpretations done by clinical labs in public repositories such as ClinVar. This is not an inevitable outcome, but the persistent work and advocacy of people like Dr. Heidi Rehm and organizations like ClinGen. We recently participated in a survey and vetting… Read more »
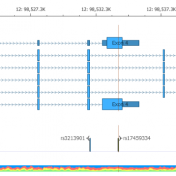
The detection and interpretation of Copy Number Variants (CNVs) is vital for the clinical evaluation of individuals with a wide range of disorders. Golden Helix has remained at the forefront of CNVs in Next-Gen Sequencing (NGS) data since 2016 with the release of VS-CNV, our solution that allows you to both detect and analyze CNVs directly from NGS data. Earlier… Read more »
Golden Helix software provides huge analytic gain in handling large-scale genomic data. For example, a number of VarSeq users run cohort projects of whole genome level data processing hundreds of millions of variants at a time. However, many of our users are running gene panel level data for custom panels related to cancer (both hereditary and somatic), autism, cardiac, and… Read more »
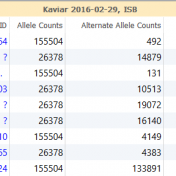
In the search for disease causing mutations it is important to determine if the variant has been previously observed in humans and at what frequency. With the advent of increasing genomic information, there is now a variety of different databases and annotation sources that can be utilized. For some, this could be a tedious task that leads only to implementing… Read more »
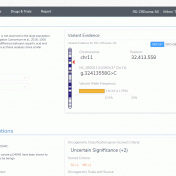
It is common knowledge that variants can be germline or somatic depending on whether the variant was inherited or acquired after birth. A well-known example is cancer-causing mutations in the BRCA genes, wherein the mutation may or may not have been inherited. Understanding the origin of the cancer-causing mutation is important when assessing potential treatment options as well as identifying… Read more »
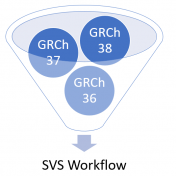
The world has been making a shift to use GRCh38 human genome reference coordinates, but the transition has not been fast. Many of the mainstay human catalog projects are changing to use native GRCh38 catalogs, or are remapping their current data to GRCh38 coordinates. While this seems to be the advancing goal, it is leaving researchers and analysts with the… Read more »
Abstract Before assessing the clinical significance of a somatic mutation, one must determine if the mutation is likely to be a driver mutation (i.e. a mutation that provides a selective growth advantage, thereby promoting cancer development). To aid clinicians in this process, VSClinical provides an oncogenicity scoring system, which uses a variety of metrics to classify a given somatic mutation… Read more »
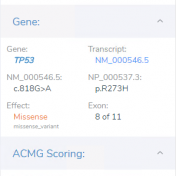
An under-appreciated area of complexity when looking into the field of genetics from the outside can be found in genes and transcripts. Alternative splicing allows eukaryotic species to have a wonderfully powerful genetic code, resulting in multiple protein isoforms being encoded in a single section of DNA. But when it comes to variant interpretation, different transcripts can result in widely different predicted… Read more »
VarSeq 2.2.1 was released on April 1st and features an upgraded gene annotation capability with new RefSeq genes tracks and an AMP workflow addition: the Drugs and Trials tab. The new RefSeq human genome genes tracks contain updated gene names and the recognition of any MANE (Matched Annotation from NCBI and EMBL-EBI) identified transcripts. VarSeq has been updated to be… Read more »
Our Support Team curates a variety of tutorials to help orient new users to the capabilities of VarSeq. We are happy to announce the team’s new release of the trio tutorial that places emphasis on using the ACMG guidelines. This tutorial gives insight into the proper setup of pedigree structure as well as detailed descriptions of the filter containers and… Read more »
Golden Helix is in a unique position to provide a secure on-premise analysis solution. This capability is based on two enablers. First, we build our software solutions from scratch and from the ground-up with the assumption that it should run on any operating system and potentially behind firewalls or even without internet access. Second, we provide these solutions on a licensing model based on training and supporting users, not… Read more »
Customizing VSClinical ACMG Guidelines Workflow Part 2 In the first part of this series, we covered how VarSeq supports customizing ACMG guidelines in VSClinical to streamline the clinical analysis workflow. VSClinical’s various customization parameters within the ACMG Guidelines workflow includes the choice of how the internal knowledge base of previous variant interpretations are stored and what considerations go into this choice…. Read more »
Clinical labs offer a unique and sophisticated product that is performed repeatedly with high standards of quality. VarSeq was developed to provide labs with the customization required for clinical genetic tests in a repeatable workflow. On top of this, VSClinical offers additional parameters and choices that can be made when designing the test workflow. In this blog series, we will… Read more »
November is the month where we pause to reflect on what we are thankful for. At Golden Helix, we are thankful for the many dedicated scientists and practitioners who are tirelessly working on new discoveries to enhance the lives of humans and preserve our food supply in the face of ever-changing conditions in agriculture. As always, we are proud to… Read more »
Our team has returned from the annual meeting of the Association of Molecular Pathology (AMP 2019), and, as always, I am grateful for all the wonderful experiences we are bringing back with us. The plenary sessions and talks were bountiful, and we were very impressed with the well-organized exhibition. Hats off to everyone involved in planning this great event! Innovation… Read more »
We have recently added a tutorial to help introduce customers to the ease and utility of the AMP Guidelines incorporated in VarSeq’s VSClinical package. The AMP Guidelines allow users to sort through available clinical evidence in a streamlined fashion to arrive at final classification and interpretation and then transfer that information into a clinical report. And the AMP Guidelines also… Read more »
VarSeq 2.2.0 was released today and this a stable release full of upgrades and polishes. Some of the newer features include the ability to store and include AMP Cancer assessment catalogs on VSWarehouse, quicker accessibility to common annotations plotted in GenomeBrowse, and the addition of all of our standard templates for the GRCh38 genome assembly. Many of the polishes were… Read more »
The common approaches to detecting copy number variants (CNVs) are chromosomal microarray and MLPA. However, both options increase analysis time, per sample costs, and are limited to the size of CNV events that can be detected. VarSeq’s CNV caller, on the other hand, allows users to detect CNVs from the coverage profile stored in the BAM file, which allows you… Read more »