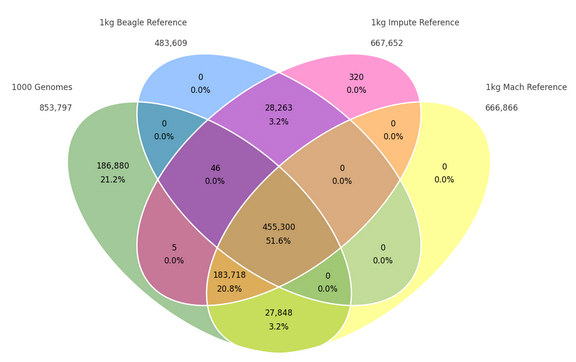
One frequent question I hear from SVS customers is whether whole exome sequence data can be used for principal components analysis (PCA) and other applications in population genetics. The answer is, “yes, but you need to be cautious.” What does cautious mean? Let’s take a look at the 1000 Genomes project for some examples.
There is a lot we can be grateful for at Golden Helix. The past year was marked by two major breakthrough launches. Earlier in 2014, we shipped SVS 8 which unified SVS with our GenomeBrowse product. We were able to improve SVS’ data management and visualization capabilities. In addition we added a number of new methods in SVS, such as SKAT-O, MM-KBAC, and various genomic prediction algorithms.
The Golden Helix team enjoys following our customers’ success. And we would like to share some recent client work to demonstrate what is possible with our software, as well as to inspire researchers to continue questioning current scientific norms.
We are shortly approaching the public launch (November 5th!) of our first clinical product, VarSeq. We could not have predicted how well the market would accept VarSeq – but we couldn’t be happier! For those of you who have not yet seen our newest product in action, I invite you to register for tomorrow’s webcast: The Golden Helix VarSeq User Experience.
Golden Helix is proud to announce the release of the Golden Helix GenomeBrowse Plugin for Ion Torrent server. The new plug-in enables adding selected BAM files from Torrent Server reports directly into GenomeBrowse. The BAM files remain on the torrent server and are streamed from the server on demand using your credentials. This feature allows GenomeBrowse users to visualize genomic… Read more »
Recently, I have been thinking a lot about Human Genome Variation Society (HGVS) notation — you know “G dot”, “P dot”, and “C dot”. HGVS has quickly become one of the most common ways to represent variants. It’s no wonder that HGVS nomenclature is used so widely. It provides an easily readable, compact representation of a variant. Since it is… Read more »
Over the last decade, DNA sequencing has made vast technological improvements. With the cost of sequencing decreasing significantly, sequencing technology has become a product for the masses. The sequencing technology and programs that were once used exclusively by major research institutions are now becoming available in many research facilities around the globe. These tools produce large amounts of data sets… Read more »
You probably haven’t spent much time thinking about how we represent genes in a genomic reference sequence context. And by genes, I really mean transcripts since genes are just a collection of transcripts that produce the same product. But in fact, there is more complexity here than you ever really wanted to know about. Andrew Jesaitis covered some of this… Read more »
We released GenomeBrowse 2.0 earlier this year, allowing users to review all types of genomic data. Since then, it has received rave reviews from thousands of users around the world. Essentially, it’s the Google Earth app for genomic data. GenomeBrowse allows a user to sift through vast amounts of genomic data, and make it easy to focus on a single part… Read more »
Up until a few weeks ago, I thought variant classification was basically a solved problem. I mean, how hard can it be? We look at variants all the time and say things like, “Well that one is probably not too detrimental since it’s a 3 base insertion, but this frameshift is worth looking into.” What we fail to recognize is… Read more »
When many people think of learning disabilities such as dyslexia and language impairment, they typically do not think of a biological or medical condition. Even more rarely do people think of these conditions as being the result of biological and genetic phenomena. However, that is exactly what I have thought of every day during my doctoral training in the Department… Read more »
Philadelphia. The City of Brotherly love, home of the Philadelphia cheesesteak, and home to the Children’s Hospital of Philadelphia, who took the No. 1 spot in this year’s U.S. News & World Report’s Best Children’s Hospitals Honor Roll. We are honored (and excited!) to sponsor this year’s Mid-Atlantic Epidemiology and Statistics (MAGES) Conference at the University of Pennsylvania. MAGES will… Read more »
Genotype imputation is a common and useful practice that allows GWAS researchers to analyze untyped SNPs without the cost of genotyping millions of additional SNPs. In the Services Department at Golden Helix, we often perform imputation on client data, and we have our own software preferences for a variety of reasons. However, other imputation software packages have their own advantages… Read more »
When researchers realized they needed a way to report genetic variants in scientific literature using a consistent format, the Human Genome Variation Society (HGVS) mutation nomenclature was developed and quickly became the standard method for describing sequence variations. Increasingly, HGVS nomenclature is being used to describe variants in genetic variant databases as well. There are some practical issues that researchers… Read more »
A few months ago, our CEO, Christophe Lambert, directed me toward an interesting commentary published in Nature Reviews Genetics by authors Bjarni J. Vilhjalmsson and Magnus Nordborg. Population structure is frequently cited as a major source of confounding in GWAS, but the authors of the article suggest that the problems often blamed on population structure actually result from the environment… Read more »
Last week Khanh-Nhat Tran-Viet, Manager/Research Analyst II at Duke University, presented the webcast: Insights: Identification of Candidate Variants using Exome Data in Ophthalmic Genetics. (That link has the recording if you are interested in viewing.) In it, Khanh-Nhat highlighted tools available in SVS that might be under used or were recently updated. These tools were used in his last three… Read more »
In a recent GenomeWeb article by Tony Fong, “Sequenom’s CEO ‘Puzzled’ by Illumina’s Buy of Verinata, Lays out 2013 Goals at JP Morgan,” Harry Hixson, Sequenom’s CEO, expresses puzzlement over why its major supplier, Illumina, is acquiring a Sequenom competitor in Non-Invasive Prenatal Testing (NIPT), and thus apparently competing with one of its major customers. In a JP Morgan interview… Read more »
Since Dr. Ken Kaufman gave his webcast on Identifying Candidate Functional Polymorphisms in SVS, we’ve been working with Dr. Kaufman to simplify and automate many of the steps in his workflow. I touched on this in my last blog post, and I’m excited to report that with Ken’s help, we’ve been able to simplify the workflow even more. In particular… Read more »
Editor’s Note by Gabe Rudy: I’ve had the chance to exchange thoughts, emails, and blog post comments for a while now with Jeff as he has written posts on NGS Leaders and engaged with me on 23andMe. He has also worked with Golden Helix software as part of Dr. Todd Lencz’s research efforts at Zucker Hillside Hospital until he recently… Read more »
Dr. Ken Kaufman’s extremely popular webinar inspired us to build new tools that would simplify the process of analyzing whole-exome DNA sequencing data even further. First I’ll describe the tools showcased in the webcast. Then I’ll detail the new tools we created to allow for a revised and simplified workflow. Subset Informative Genotypes by Category The Subset Informative Genotypes by… Read more »