In the previous two articles, we explored the different steps of a clinical workflow. The first post covered the automated analysis that creates a VarSeq project. While the second post covered the interpretation steps and generation of a clinical report. These posts illustrated the ease with which these complex tasks can be carried out. Today we’ll dig a little bit deeper to see the steps and configuration that make this such an easy and repeatable task.
The workflow is broken into a couple different scripts based on the software that is used. In this example, the first script watches for the presence of the input files. When all the inputs are present it starts a second script to process this batch of samples. The batch script handles the automated steps that we covered in part one of this series. Once this script is complete we will have the completed VarSeq project which we can pass off to the lab tech to start the process outlined in part two of this series.
Watch Script
The watch script is a simple bash script. It watches a predetermined input directory. When the files in the directory change it checks to see if all of the required files for the pipeline script are present. The script can be set up as a service or daemon that continually watches the input directory. If the watched directory is available as a shared drive on the local network, the sequencer can copy the data to drive as soon as it finishes the run. The only other required input is the sample information.
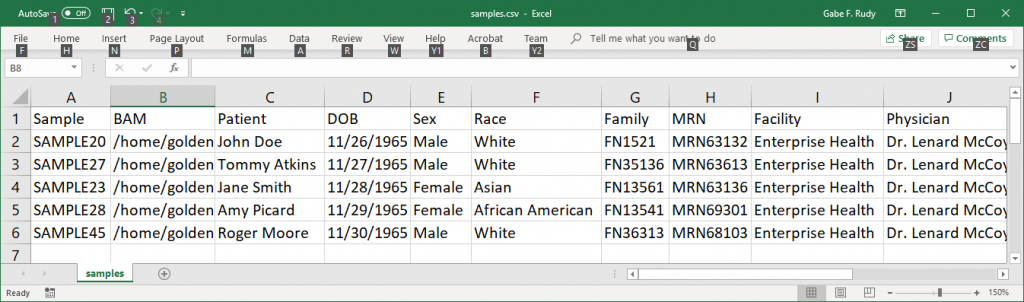
The sample information is passed into VarSeq as a CSV text file. The sample information may include phenotypes or target genes to do phenotype based gene ranking or other parametrization of the VarSeq project. Additionally the sample information may include patient details such as the diagnostic tests performed, the ordering physician, and medical record numbers. This is used to fill in all of the relevant details in the sample report. To automate this step the information can be pulled from an EMR or LIMS system. Otherwise it will need to be created by hand. The only other required inputs are the BCL or FASTQ files from the sequencer. After all the prerequisites are complete, the watch script will start the pipeline script with the inputs.
Pipeline Script
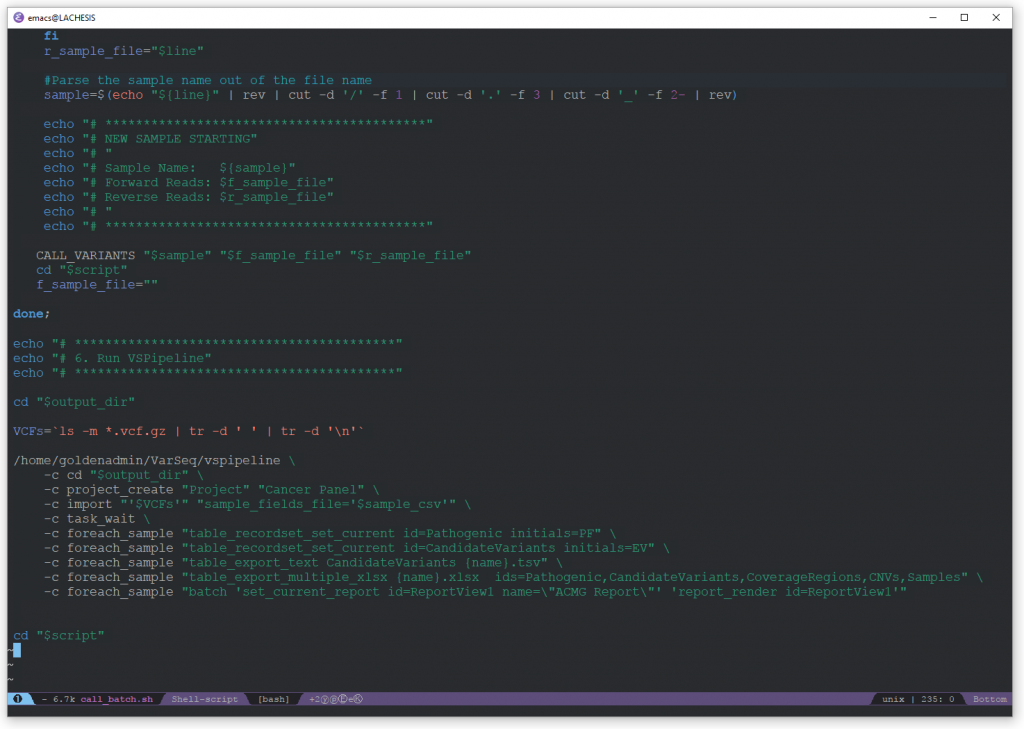
The pipeline script takes the input and output directories as arguments. From the input directory it selects the input FASTQs based on a predetermined naming convention. These inputs are then threaded through the Sentieon pipeline steps of alignment, deduplication, BSQR, indel realignment, and variant calling. Each of these steps has it’s own command in the bash batch script. Since each command is separate, each one has it’s own set of parameters which can be adjusted. This allows for fine tuning of the pipeline when it is being set up and validated. The final step of the script calls vspipeline. This combines the VCFs and BAMs from Sentieon, the sample attributes file, and the VarSeq project template to create a VarSeq project. This step calls CNVs and runs all of the desired annotations and algorithms so that the created project is ready for the lab tech to start QC analysis. This finalized project can be copied to a shared network location so that the lab tech can easily find and open it on their PC.
Putting it All Together
Creating the pipeline scripts and setting them up so that they run automatically is a complex task. It requires Linux administration and bioinformatics experience, as well as an understanding of the VSPipeline and Sentieon tools. While all the tools are well documented, it can be a significant investment to get up to speed on all the different parameters and their nuances. Therefore, we provide setup and installation as a service to help our customers get up to speed quickly and efficiently. If you would like help setting up your automated pipeline don’t hesitate to reach out. Setting up all the scripts to run automatically is a huge win for the lab. It means that the day to day users of the pipeline do not need to have any command-line experience. Instead, they can start the sequencer with the comforting knowledge that a VarSeq project is on the way.