Probably one our most popular public annotation sources we curate and update is the database of Non-Synonymous Functional Predictions (dbNSFP). In it’s recent update, it has expanded the predictions to include FATHMM-MKL and VarSeq now incorporates this new prediction into its voting algorithm of now 6 different discrete predictions per variant.
You can update to dbNSFP 3.0 using the built-in ability to move to the latest annotation in VarSeq 1.3.1 or later. Just click on the “information” icon on your variant table to see available updates and select one or all of them.
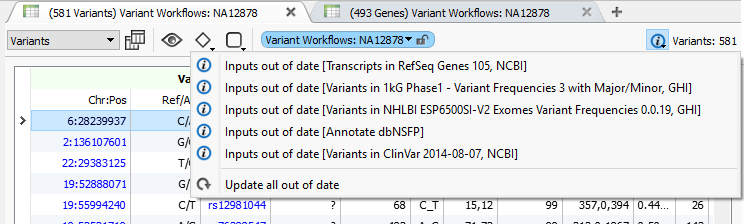
With VarSeq 1.3.1, we added a notification that allows you to easily update one or all of your out-of-date annotation sources to the latest published version.
Since its publication in 2011, dbNSFP has been faithfully updated to incorporate not only functional predictions, but also other conservation scores and combined machine learning based scores such as VEST3 and MetaSVM on every non-synonymous variant in the human genome.
Because of the extent of these scores, we provide two versions of the annotation for every release:
- dbNSFP Functional Predictions 3.0, GHI: A subset of only 17 fields providing discrete predictions on variants. We provide friendly names for these predictions, such as Tolerated, Damaging etc, versus the dbNSFP encoding of “T”, “D”.
- dbNSFP Functional Predictions and Scores 3.0, GHI: The full set of 69 fields, including all the raw and ranked-scores supporting the discrete predictions, as well as conservation and combined/meta scores.
In this release, there are 82.8 million individual predictions, yet the 17-field downloaded annotation requires only 411MB!
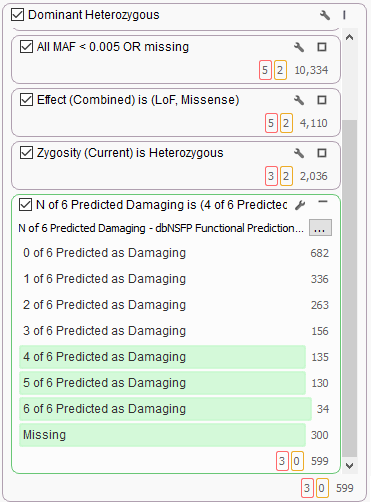
It’s common to use dbNSFP to help find variants of interest in the broad search for candidate dominant model heterozygous variants of functional significance. Here we see the new 6-algorithm voting in dbNSFP 3.0 helping to stratify 2,000 candidate variants and filtering only majority-voted variants.
The 3.0 release of dbNSFP added FATHMM-MKL prediction algorithm and the fitCons conservation score.
Although it shares the same name as FATHMM, the new FATHMM-MKL is comparable to CADD and GWAVA in its ability to predict the functional consequence of coding and non-coding variants. In its recent publication, they outline their method of feature selection (picking from regulatory, conservation, GC content, TFBS and other “features” about the genomic context of an allele substitution) as well as their multiple kernel learning (MKL) method that combines many Support Vector Machines (SVM) with weights determined by the informative-ness of different data types and performance on training sets.
After doing some evaluation of the FATHMM-MKL predictions versus the current SIFT, Polyphen2, Mutation Taster, MutationAccessor and FATHMM algorithm, I confirmed that it provided an independent classification not strongly correlated with any of the existing classifiers for variants that are split in their voting (2 or 3 out of the existing 5 predicted a variant Pathogenic).
For this reason, we updated our annotation algorithm to include it as a 6th voter, meaning there can now be up to 6 out of 6 algorithms agreeing that a variant is Damaging or Tolerated.
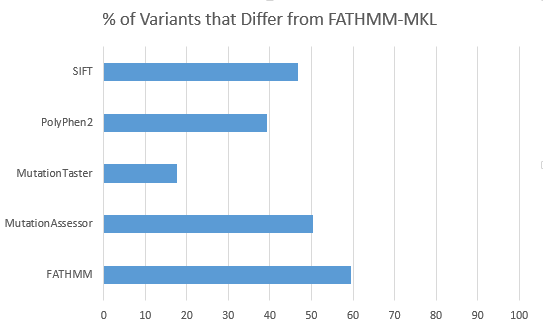
For 2,632 variants from a filtered exome that were previously “split” in their functional prediction with 2 out of 5 or 3 out of 5 of SIFT, PolyPhen2, MutationTaster, MutationAssessor and FATHMM predicting the non-synonymous variant to be “Damaging”, FATHMM-MKL differs the least in its own prediction with MutationTaster and differs the greatest with FATHMM.
While this is one of the most challenging public annotation sources to curate from its raw 76GB chunks of text, through the many edge cases and transformations to get to the highly accessible and documented form we strive for in all our annotations, it is also one of the most powerful in its ability to enhance the variant interpretation workflow and user experience.



