When you think about the cost of doing genetic research, it’s no secret that the complexity of bioinformatics has been making data analysis a larger and larger portion of the total cost of a given project or study. With next-gen sequencing data, this reality is rapidly setting in. In fact, if it hasn’t already, it’s been commonly suggested that the total cost of storing and analyzing sequence data will soon be greater than the cost of obtaining the raw data from sequencing machines.
In my previous post, A Hitchhiker’s Guide to Next Generation Sequencing – Part 1, I covered the history and forces behind the decreasing cost curve of producing sequence data. Through innovation and competition, high throughput sequencing machines are making it more affordable to use whole exome or whole genome sequencing as a research tool in fields that benefit from deeply understanding the genetic components of disease or other phenotypic traits.
In this post I plan to explore, in depth, what goes into the analysis of sequence data and why both the cost and complexity of the bioinformatics should not be ignored. Whether you plan to send samples to a sequencing-as-a-service center, or brave the challenges of sequencing samples yourself, this post will help distinguish the fundamental difference in analyses by their usage patterns and complexity. While some bioinformatics packages work well in a centralized, highly tuned and continuously improved pipeline, others fall into a long tail of valuable but currently isolated tools that allow you to gain insight and results from your sequence data.
Breaking Down of Sequence Analysis
The bioinformatics of sequence analysis ranges from instrument specific processing of data to the final aggregation of multiple samples into data mining and analysis tools. The software of sequence analysis can be categorized into the three stages of the data’s lifecycle: primary, secondary, and tertiary analysis. I will first define these categories in more detail and then take a look at where the academic and commercial tools currently in the market fit into the categorization.

Primary analysis can be defined as the machine specific steps needed to call base pairs and compute quality scores for those calls. This often results in a FASTQ file, which is just a combination of the sequence data as a string of A, C, G and T characters and an associated Phred quality score for each of those bases. This is the absolute bare minimum “raw” format you would ever expect to see for sequence data. While the first generation of high throughput sequencing machines, such as the Illumina G1, allowed for users to provide their own alternatives to the standard primary analysis solution, called “the Illumina pipeline”, current generation machines often do this work on bundled computational hardware. The effective output of the machine is the result of the primary analysis. This output is ready for processing in a secondary analysis pipeline.
Because current sequencing technologies are generally based on the “shotgun” approach of chopping all the DNA up into smaller molecules and then generating what are referred to as “reads” of these small nucleotide sequences, it’s left up to secondary analysis to reassemble these reads to get a representation of the underlying biology. Before this reassembly, the “raw” reads from the machine are often assessed and filtered for quality to produce the best results. Reassembly differs if the sequencing was done on an organism with a polished reference genome or if the genome is to be assembled from scratch, also referred to as de novo assembly. With a reference genome available, the process is much simpler, as the reads simply need to be aligned to the reference, often with some tolerance for a few base-pair errors in the sequences of the reference itself.
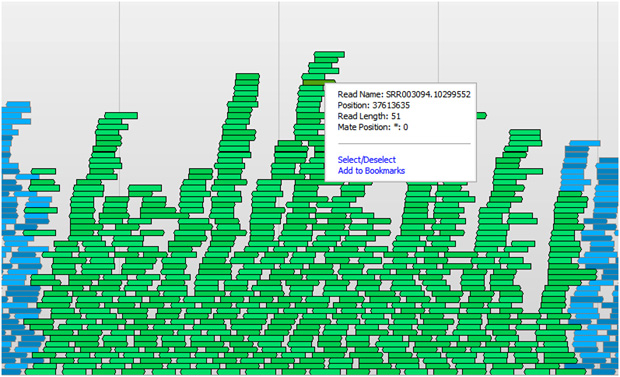
Pileup from the Savant Genome Browser
In both the case of de novo assembly or reference sequence alignment, you will be shooting for a desired average depth and coverage of sequence reads over the entire genome or targeted regions of interest. Depth is a measure of how many reads cover a given locus of the genome. If you were to pile up the reads to where they were assembled or mapped (pictured above), the depth would be the height of this pileup at each locus. For de novo assembly, a higher average depth is usually needed, so that large contigs can be formed that are then the building blocks for a draft genome. In the case of sequence alignment, higher average depth means more certainty in the “consensus” sequence of the sample and more accuracy in detecting variants from the reference.
The next step in the sequence analysis process is detection of variants. While more customizable, and sometimes considered part of tertiary analysis, variant calling lends itself to being pipelined in the same manner as secondary analysis. Variant calling is the process of accurately determining the variations (or differences) between a sample and the reference genome. These may be in the form of single nucleotide variants, smaller insertions or deletions (called indels), or larger structural variants of categorizations such as transversions, trans-locations, and copy number variants.
Tertiary analysis diverges into a spectrum of various study specific downstream investigations. Though the research or building of draft genomes for novel variants will have its own specialized tertiary analysis after de novo assembly, I will focus on the more mainstream practice of “resequencing” studies where sequence alignment to a reference genome was used. Out of the secondary analysis step of variant calling, you now have a more manageable set of differences between the sequenced samples and the reference, but there is still an enormous amount of data to make sense of. This is the realm of tertiary analysis.
The Resource Requirements of Primary and Secondary Analysis
As I described in Part 1, the amount of data produced by current generation, high throughput sequencing machines is enormous and continues to grow. Primary analysis solutions are largely provided by the platform providers as part of the machine’s function. Whether you run their software on bundled compute resources or your own cluster, the analysis is designed to keep up with the throughput of the machine as it produces measurement information, such as images of the chemistry. In contrast, secondary analysis performs operations on the aggregated data from one or more runs of the machine. As a result, secondary analysis requires a significant amount of data storage and compute resources.
Usually quite resource intensive, secondary analysis performs a given set of algorithms and bioinformatics on a per-sample basis. This repeatable process can then be placed in an analytic pipeline that’s entirely automated. Such an automated pipeline allows for even more resource utilization through scheduling. It also allows for a process of continuous improvement of the pipeline to be employed by monitoring quality metrics and tweaking or improving the algorithms and their parameters. Although these pipelines may be nothing more than an amalgamation of single purpose tools (as we will see in the next section), the economies of scale, not only in resource utilization but also in expertise and quality improvement, are realized by having secondary analysis centralized to a core lab or sequence-as-a-service provider.
Current Software Solutions
Over the past few years, the methods and algorithms designed for the primary and secondary analysis of high throughput sequence data have matured to satisfy the common needs of analysts. Most of these methods can be found in open source, single-purpose tools that are freely available to use and modify. In the case of de novo assembly, Velvet, written by Daniel Zerbino at EMBL-EBI uses de Bruijn graphs for genomic assembly and has set the bar for quality and accuracy. For sequence alignment to a reference genome, various Burrows-Wheeler Alignment based algorithms achieve a great balance of speed and accuracy. The original paper describing this use of the BWA algorithm is implemented in the aptly named BWA package, a successor to the gold standard, but slower MAQ. Other BWA implementations include bowtie and SOAP2.
Seemingly simple in nature, a lot of work has gone into accurate variant detection for resequencing data. Single Nucleotide Variant (SNV) detection has gone through a few generations of algorithmic improvements, with implementations such as SOAPsnp and GATK representing the current lineup. Indel detection, being a newer pursuit, has not had the time for a real winner to emerge. Complete Genomics has shown that a more holistic approach may achieve better results than current best practices. Finally, copy number and other structural variant detection are still being actively developed as new methods. These types of analyses require new ways to handle the unique properties of sequence data.
Though the above methods are often implemented in single-purpose packages, they can be strung together to compose a state-of-the-art secondary analysis pipeline. This can be done manually with a considerable amount of IT customization, or through a server-oriented connector package such as Galaxy from Penn State. Commercial offerings in this area, such as CLC bio, attempt to provide an all-in-one solution for secondary analysis with their own proprietary tweaks on the standard algorithms, and a GUI for running a single sample at a time through each step.
Tertiary Analysis Requirements
After getting sequence sample data to the stage of called variants, the real work begins in making sense of the data in the context of your study. At this point, multiple samples need to be brought together, along with phenotype and other experimental data. While primary and secondary analysis can be automated and centralized, here in the “sense making” stage, there are a plethora of potential different analysis techniques and exploratory paths. Fortunately, even whole genome sequence data processed to the level of variant calls is manageable on a researcher’s modern desktop. Whole exome or targeted resequencing is even more manageable.

Unlike microarray data, where the probes are usually carefully selected to be loci of potential interest, sequence data naturally contains variants regardless of their loci or functional status. As illustrated in the categorization of operations above, the common starting point in tertiary analysis then, is to use all the annotation, functional prediction, and population frequency data available to sort through this unfiltered list. Luckily, the genetics community has really stepped up to the plate in both funding and sharing public data repositories. Population cataloging efforts such as HapMap and the 1000 Genomes Project allow for studies to be able to determine the relative frequencies of variants in their samples compared to common populations. Once variants have been filtered and ranked, new methods are gaining momentum to handle the analysis of rare variant burden as a model of disease association, as well as other methods specific to the filtering and studying of sequence data.
Common also in the exploratory analysis of sequence data, is the use of a genome browser to give historical and biological context to your data. Like these other filtering and analysis methods, a genome browser will utilize the many repositories of genome annotations and public data. You may want to see if a given locus has had GWAS results published in the OMIM database, compare variants against repositories of known SNPs, or just visualize the genes from various gene catalogs for a given region.
Conclusion
As we can now see, there’s quite a bit to consider in terms of next-gen sequencing analysis, from producing raw reads to calling variants to actually making sense of the data, all of which can add to the complexity and cost of a sequencing study. As the field standardizes on best practices and methods for primary and secondary analysis, we can be more certain about planning for these steps and even include them in the “cost of production” of sequence data from either your own core lab or a “sequencing-as-a-service” partner.
Tertiary analysis is still in its infancy but as we experienced at IGES and GAW this year (see our report), promising new methods and workflows are beginning to emerge. Over the coming year, we are probably as excited as you are to see breakthrough discoveries being made with this technology. In my third and final post of this series I will be exploring tertiary analysis in more depth focusing primarily on genotypic variants: the current state of sense making, how the field is progressing, and the challenges that lay ahead.
…And that’s my two SNPs.
Pingback: Tweets that mention A Hitchhiker’s Guide to Next Generation Sequencing – Part 2 | Our 2 SNPs… -- Topsy.com
Pingback: Our top 5 most visited blog posts | Our 2 SNPs…®