I’m sitting in the Smithsonian Air and Space Museum basking in the incredible product of human innovation and the hard work of countless engineers. My volunteer tour guide started us off at the Wright brother’s fliers and made a point of saying it was only 65 years from lift off at Kitty Hawk to the landing of a man on the moon.
I was dumbstruck thinking that within a single lifetime the technology of flight could go from barely existing to space travel.
I can’t help but think about the parallel journey we are on that started with the discovery of DNA’s molecular structure by Watson and Crick. The technology and scientific advances since then are similarly staggering.
Given that discovery was 52 years ago, we only have 13 more years to achieve our moon shot! That is more time than between JFK setting the deadline of a manned mission to the moon and the first lunar landing in the summer of 1969, but I hope we can have our own JFK goal-setting moment soon.
Given an intro like that, you may think I’m about to announce a moon shot. Well, I might as well disappoint you now. Although I’m excited that we are making our own small step this year, and I thank those of you who have been talking to us and are stakeholders on that project, I’m going to cover something more modest today.
While the astronauts were enabled by rocket engines and carefully computed lunar orbit-reaching trajectories, the modern Cromonaught needs usable bioinformatics tools and lots of shared community-generated data to advance the frontier.
The best tools would be as effective as an aimless rocket without this data, so it’s something we invest in heavily.
As I illustrated in my last blog post, Golden Helix takes our role in curating the community-driven repositories of data into a versioned and usable form seriously. Once converted and documented, we published the latest version of a data source in perpetuity to our free public annotation repository, ready for visualizing in GenomeBrowse or analysis in SNP and Variation Suite.
Over the last couple of months, we have been focused on crafting the best possible versions of some of the core data sources used by Cromonaughts: ClinVar and dbSNP.
ClinVar: The International Space Station of Data Sharing
Through the visionary efforts of Heidi Rehm and others, the walls on lab-specific data silos are slowly coming down as the prospect of having a comprehensive repository for sharing clinical annotations of variants and disease association overcomes the inherit lab interests to keep costly classification work privately held as a competitive advantage.
While the NCBI, as part of its ownership of ClinVar, does monthly VCF dumps of its database, the file encodes the important fields in compact and opaque forms.
Compounding this, the VCFs pack all ClinVar records for a given chromosome position onto a single line (record). Because ClinVar has different pages for the classification of a variant against different diseases, this can result in heavily overloaded fields.
You can see this as a comparison of our previous and latest ClinVar annotation at this splice site SNP in the BRAF oncogene:
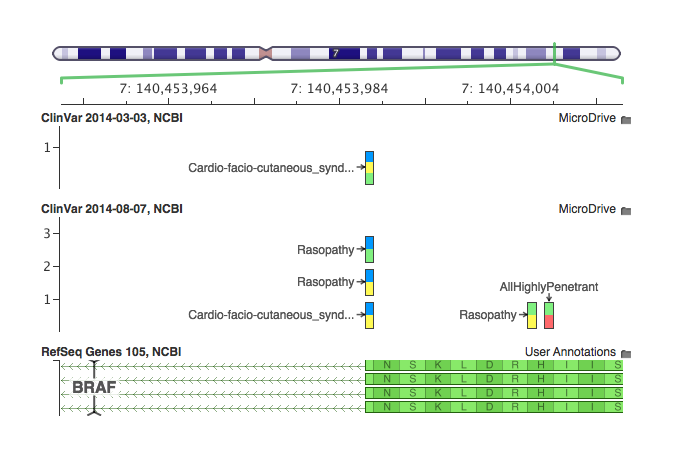
Our previous curation of ClinVar was a direct conversion from the NCBI VCF file. You can see there is a single multi-allelic record. That record contains data from three distinct ClinVar variants: phenotype pairs – each one has it’s own page with a different clinical classification, history of evidence, and review status (the stars providing certainty of the classification).
With our new conversion script, we create one record per ClinVar entry, showing that there are two distinct alleles classified as Pathogenic for Rasopathy, and one classified as Pathogenic for Cardio-facio-cutaneous syndrome. This not only makes the visualization clearer, but greatly improves the ability for the annotation and filtering process to be more precise in specifying classifications or disease names and keeping relevant Accession IDs and other ClinVar attributes together.
Secondly, the reason I know those variants are Pathogenic is that I can now click on each record and see the well formatted fields:
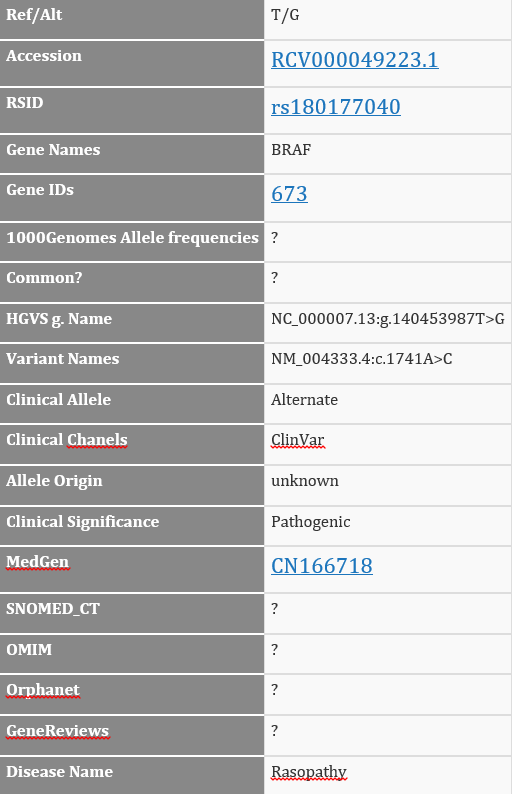
In comparison, here is what the previous direct-from-VCF version looked like:
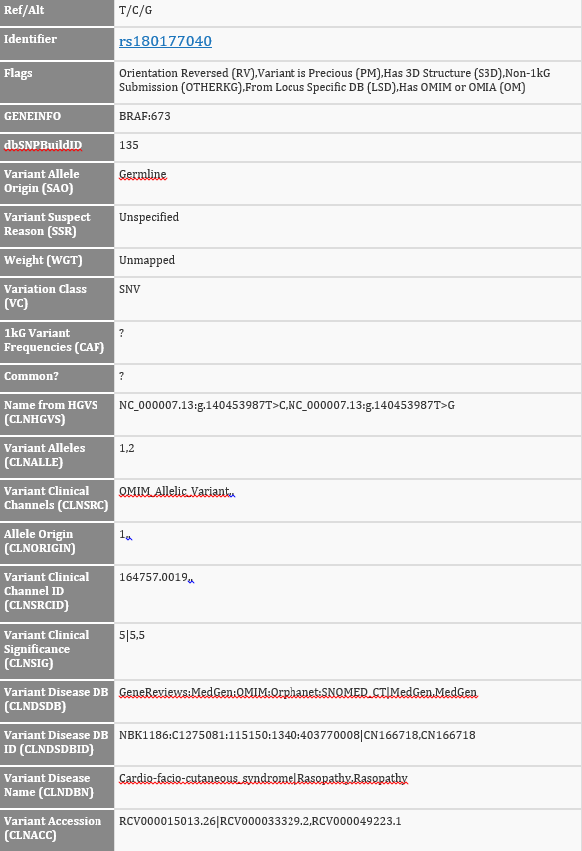
In the raw VCF file, the CLNSIG field with the pathogenic information for these three records has the value of “5|5,5”. You can certainly figure out what the two types of delimiters mean, and that 5 maps to “Pathogenic”, and then track which of the alleles and records to which each classification refers, but now you don’t have to!
Closing the Gap of NGS and Historical Representations
If you have attended one of my short courses or webcasts focused on public data sources and variant annotation (like the one I just gave at the Clinical Dx Summit here in DC), you may have heard me harp on the different representation of the same variants.
The example I frequently use, because of its weighty implications, is the Delta F508 mutation in CFTR that is the most frequent carrier mutation in Caucasians and, inherited recessively, will result in a Cystic Fibrosis diagnosis with 100% penetrance.
A couple years ago, a customer pointed out that their internally curated representation of CFTR2 (the FDA recognized database clinically assessed variants for CFTR) did not match their gene panel variants being left-aligned by standard variant caller convention.
We came up with a solution at the time for their specific database, but it has bothered me ever since that this is just one of potentially many InDels that we could be mis-annotating due to their multiple representations as DNA variants.
Clinical databases often inherit their definition of a variant from the way it was described in historical assays, which can differ from the NGS standard of placing insertions and deletions (indels) in their left-most (5′most) representation. In fact the HGVS standard for representing neucleotide indels variants is to use the rightmost (3′most) representation.
This can sometimes be a non-obvious transformation as these two representations of this identical insertion demonstrate. It’s a fun exercise to write out the alternate sequence using the reference to see how they equate.

In fact, to properly left-align variants the best approach is to use the full power of a local Smith-Waterman alignment algorithm between the reference and alternate sequences with some flanking window.
Following the lead of vcflib, the command line Swiss Army Knife of variant manipulation, we added the ability to do a SW-based left-align in our GenomeBrowse convert wizard so we can left-align sources like ClinVar!
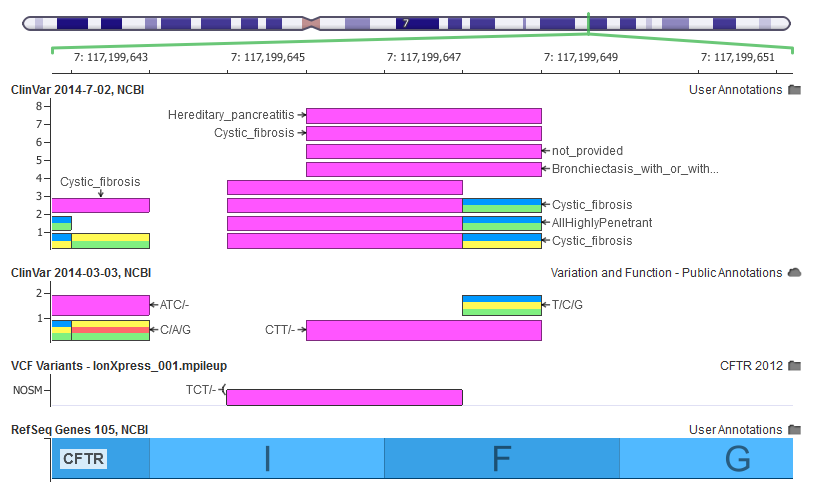
Back to my favorite example, on the bottom you can see where this variant is called with a NGS pipeline in a VCF file. Above that the previous ClinVar has it 1 bp to the right and would not have been annotated by a position match. Finally we have our new ClinVar, which first breaks out the record into the four different disease classifications ClinVar provides, and then those four records are copied to the left-shifted position to exactly match the VCF variant.
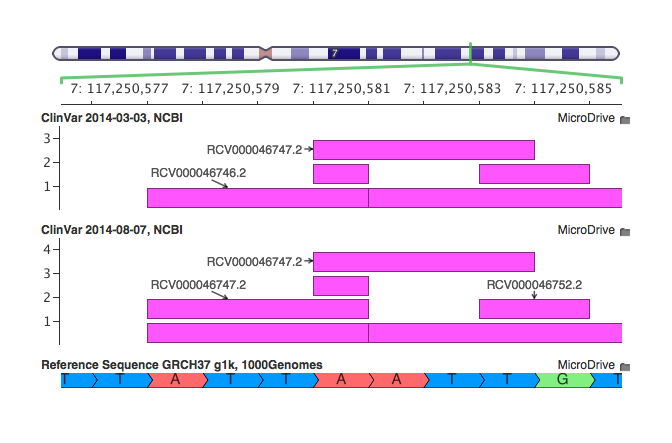
Here is one final example where you can see that a variant (RCV000046747.2) on the top graph is, in fact, the same mutation cataloged under RCV000046746.2 and matches it when left-shifted. In this case ClinVar has two records for the same physical mutation. This is to be expected when data is collected from different sources without a vigorous normalization algorithm. In fact dbSNP has DeltaF508 in both of the representations above with different RS ids.
dbSNP 141: Launching the Saturn 5
If it sounds like I’m being harsh on NCBI, I’m really not. They do an amazing job being the hub of so much data and have a responsive support team that cares about making their data releases useful and accurate. I actually asked this week for the Review Status field of their database (the prominent star rating at the top of the page) to be added to their VCF outputs and they assured me future releases will have this.
From what I can tell, the recently released build 141 of dbSNP was a massive effort. It is the first build to map to both the GRCh37 and GRCh38 human references. I can imagine getting it shipped was a bit like moving the 6.5 million pounds of a Saturn 5 with 7.5 million pounds of rocket engine thrust.
Soon after their announcement, we got to work curating and validating this release. As you might see in NCBI’s notes, they did some clean up of records over previous release. So I did my own comparison analysis with build 138 by looking at variants in some public exomes where multiple dbSNP records mapped to the same site, and there is an obvious improvement (dbSNP 138 had a number of surperfluous records that have been cleaned up).
But then I saw this:

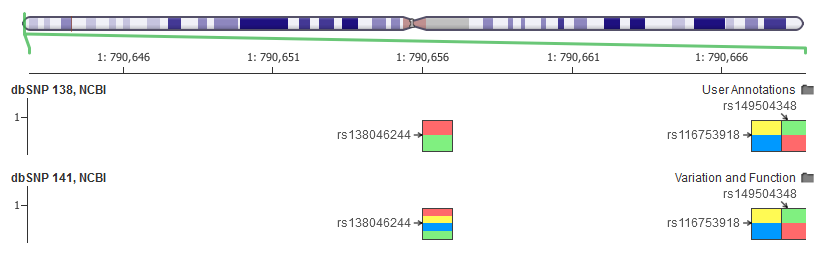
The SNP on the top of the 141 track, rs386523011, has 4 alleles present in the VCF and thus is rendered with the four allele colors in GenomeBrowse, but if you click through to the dbSNP page, it says it’s a G/A SNP like the others here.
Compared to 138, there were thousands of sites in my exome analysis where the dbSNP entries had four alleles listed rather than the two in the official database.
I did some more manual sifting through the VCF and found this example that wasn’t just at a site with multiple clustered SNPs.
Sending this over to the dbSNP support team, I got a quick response and after a couple back and forths they confirmed the issue and coded up a fix to their VCF creator that fixed the issue.
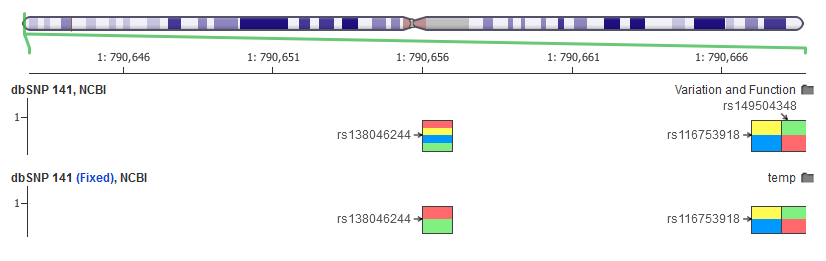
So we now have curated the corrected dbSNP 141 and also left-shifted it just like ClinVar (yes, that takes a long time!) to allow you to easily annotate against all records for a given mutation. [Note that we are waiting for the “official” VCF to be updated before publishing 141]
Enabling Regular Updates
With each custom curation task my team tackles, we make sure to set ourselves up for a very quick turn-around when these sources have new releases. ClinVar has monthly releases, and dbSNP tends to have a new build yearly.
More importantly, each version can be downloaded and used as your “frozen” source for analysis. You can even see every previous version of a given source by un-checking the default “Latest” filter in the Annotation Library window.
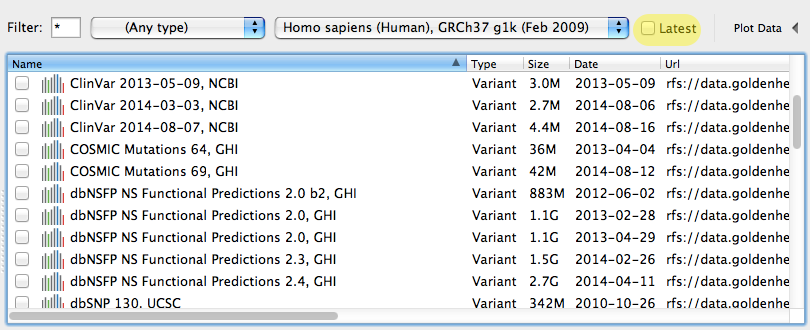
Unlike the single integrated database approach that many cloud based variant annotation vendors go for, this architecture allows us to keep up with the latest public data sources and allows users to have complete control of when and to which version they update their analysis workflows.




2 Comments