
In a recent blog post, we explored how phased genotypes provide crucial insights by separating variants into distinct haplotypes—groups of alleles inherited together from a single parent. We also discussed how the combined impact of multiple variants within the same gene can significantly differ from their individual effects. However, accurately assessing the joint impact of these in-phase variants is a challenging task, requiring specialized analysis tools.
In this post, we introduce a new algorithm in VarSeq that merges nearby in-phase variants, allowing for the analysis of their compound effect.
Algorithm Overview
The Collapse Phased Variants algorithm merges close proximity variants that are in-phase, using different thresholds for substitutions and length-altering variants:
- For substitutions, variants are merged if they are within three base pairs of one another.
- For length-altering variants, the distance threshold is a user-defined parameter with a default value of 100bp.
When two or more variants are determined to potentially result in a compound effect based on their proximity and shared phase, the algorithm merges them into a single-phase collapsed variant. The algorithm then computes a new genotype value for each collapsed variant based on the genotypes of the merged individual variants.
Example 1: Complex Substitutions
Let’s look at an example where the algorithm can be leveraged to reveal the compound impact of two nearby substitutions. In the screenshot below, we see the output of the Collapse Phased Variants algorithm. At the bottom of the window, we have a split table view with phase collapsed variant on the left and the original unmerged variants on the right.

When considered independently, the two original variants imported from the VCF are predicted to be missense mutations with uncertain impact on protein function. However, by applying the Collapse Phased Variants algorithm, we can assess their combined impact, revealing that these mutations together create a stop-gain, likely triggering nonsense-mediated decay. This combined effect can be visualized in GenomeBrowse, where the phase-collapsed variant is plotted below the original variants, providing a clear view of their joint impact.
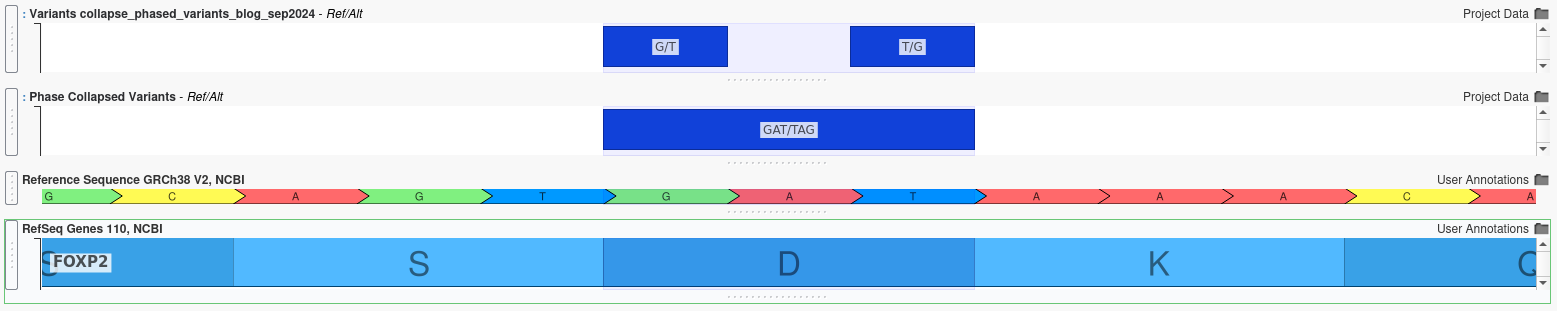
Example 2: Length-Altering Variants
When multiple insertions and deletions occur within the same gene, their combined effect can differ dramatically from their individual effects, even if the variants are not in close proximity. For example, the two variants shown below are separated by several amino acids:

Individually, each variant appears to cause a frameshift mutation, which would be likely have a severe, deleterious effect. However, when analyzed together using the Collapse Phased Variants algorithm, it becomes clear that the reading frame is preserved, potentially mitigating the individual effect.
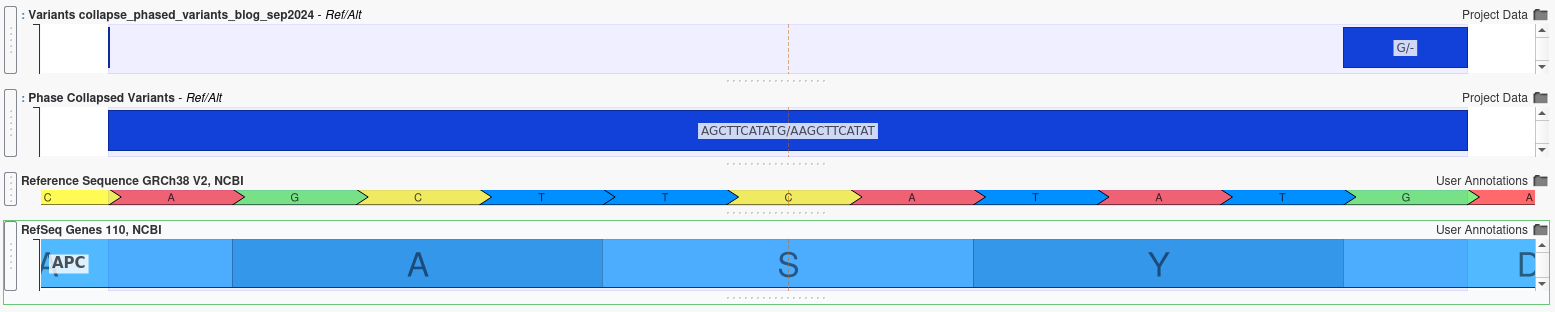
Analyzing Collapsed Phased Variants
The merged variants produced by the Collapse Phased Variants algorithm can be annotated, filtered, and interpreted just like any imported variants in VarSeq. In the screenshot below, we’ve applied several quality filters to our Phase Collapsed Variants table, along with a filter based on the automatic classification provided by our ACMG Classifier.
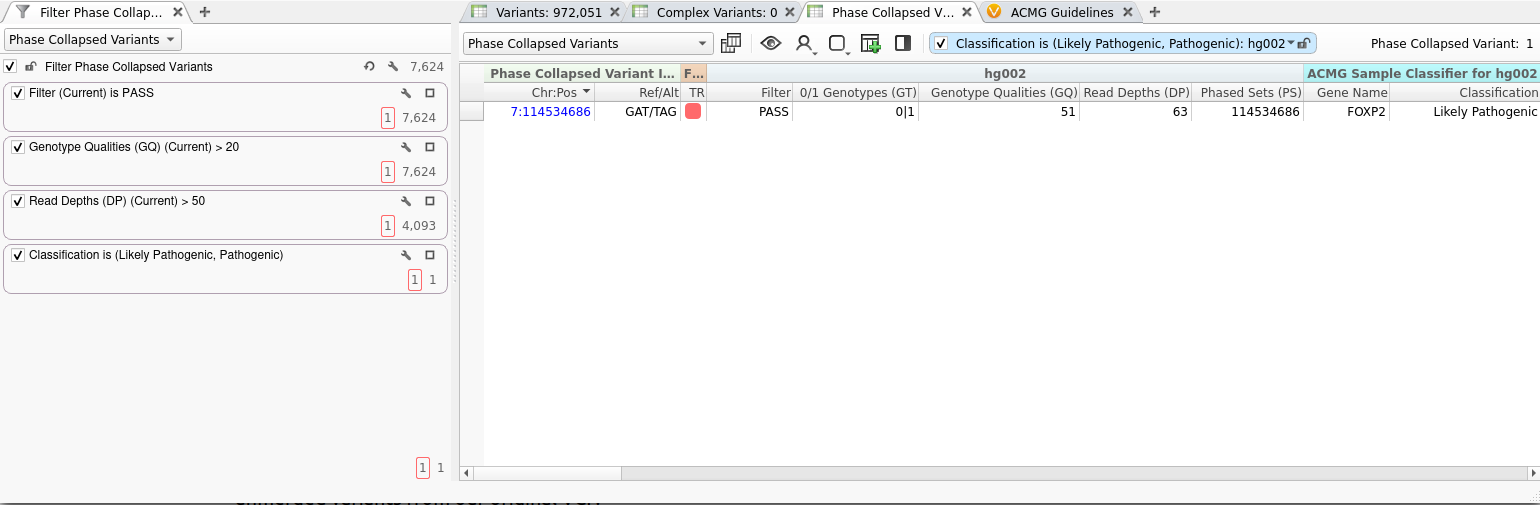
Although the algorithm generated thousands of phase-collapsed variants, our filter chain has identified a single likely pathogenic variant, which we’ve prioritized for manual interpretation. Using the split view discussed above, we can easily examine the constituent variants that were merged to construct this phased collapsed variant.

After reviewing the variant in VarSeq, we’re ready to move forward with manual interpretation using the ACMG Guidelines workflow in VSClinical.
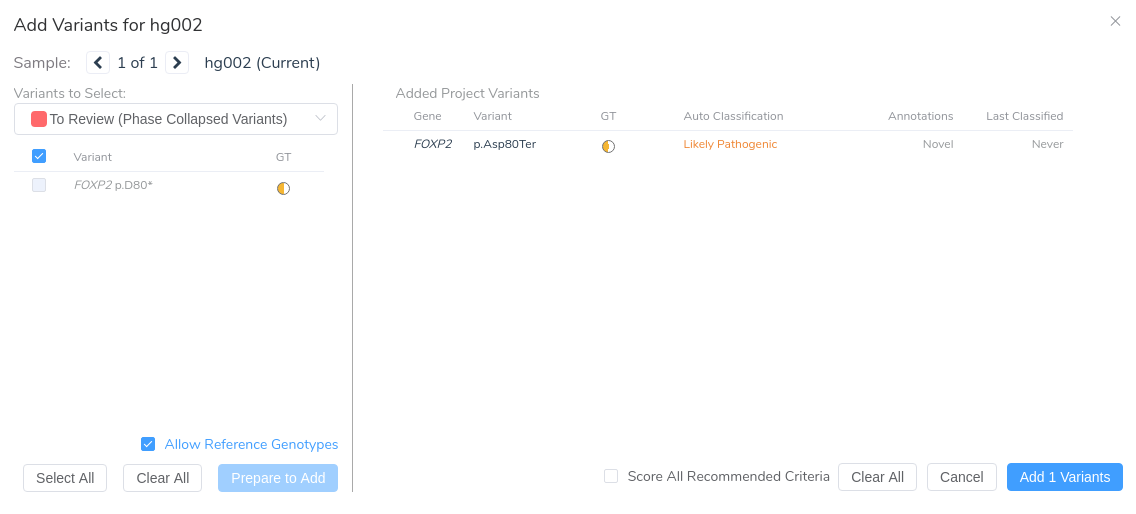
Once the variant is added to our VSClinical evaluation, we can assess the relevant criteria, interpret the variant according to ACMG Guidelines, and include it in the clinical report alongside any significant unmerged variants from our original VCF.

Conclusion
VarSeq’s new Collapse Phased Variants algorithm offers a powerful tool for assessing the joint effects of nearby genetic variants. This algorithm’s ability to construct complex variants and visualize them in VarSeq enables more accurate functional predictions and prevents misinterpretation of variants that would otherwise be interpreted independently.
If you are interested in incorporating this powerful tool into your NGS analysis, please reach out to us at [email protected].



