
With the release of VarSeq 2.5.0, Golden Helix customers can now perform an unprecedented number of workflows within a single software suite. Carrier status analysis, multi-sample clinical workflows, and a built-in oncogenicity scorer augment the already diverse and robust set of tools encompassed by VarSeq. In addition, we have continued to improve VarSeq’s ability to handle multiple data inputs, from SNPs and indels to CNVs and SVs. Paired with improved speed and expansive tools for automation, VarSeq is better equipped than ever to handle the needs of modern clinicians working in the NGS space, from up-and-coming labs to massive genome centers.
Of course, VarSeq is only part of the picture, and a tertiary analysis tool is only as good as the data fed into it. While VarSeq supports data generated with virtually any tool that can produce a VCF (for SNPs/indels, CNVs, and SVs), alignment file (BAM or CRAM), or formatted genomic signatures (e.g., TMB, MSI, etc.), our partnership with Sentieon enables VarSeq users in need with a fast and precise secondary analysis tool. We’ve previously highlighted some exciting Sentieon benchmarks, and Sentieon’s offerings continue to grow. We are excited to expound upon some of the new features that our partners at Sentieon have been hard at work developing.
Exciting updates from Sentieon
Sentieon’s last couple of updates have been big ones. Potentially the most impactful changes have been the improvement of Sentieon’s cutting-edge machine-learning model files that improve the speed and accuracy of their flagship variant callers. In an exciting speed update, they’ve added this capability to the BWA-MEM algorithm as well, allowing users to incorporate a model file during alignment and significantly increase the overall secondary analysis pipeline’s throughput.
Improving our secondary analysis bundle
To better serve our users, we’ve incorporated these changes into our shipped starter scripts. We’ve also updated our shipped scripts to match Sentieon’s best practices, allowing them to serve as a robust starting point. We expect that most of our users employing Sentieon for the secondary analysis will need to refine these pipelines to meet their needs, and hence seek only to provide the relevant tools, and not necessarily a complete solution. That being said, we’ve expanded how those tools are used in a couple of simple ways while keeping everything streamlined and familiar.
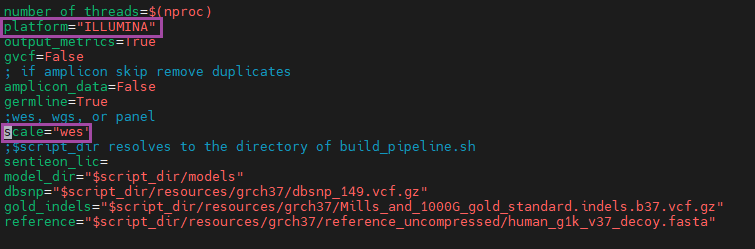
For those who are unfamiliar with our bundled Sentieon deployment, our secondary analysis documentation is a great place to start. On the other hand, current users can run the update_scripts.sh script to pull our latest improvements and access some of the new scripts leveraging Sentieon’s model file updates. From there, virtually all of the changes can be handled through the familiar initiation files, variant_calling.ini and variant_calling_38.ini (Figure 1). Here, we’ve added fields to define the scale of data (gene panels, exomes, or whole genome sequencing) and the sequencer used to produce the input FASTQs. Presently, users who select "ILLUMINA" for the sequencer can generate variant-calling scripts, including the new model files.
Making full use of Sentieon’s broad support resources
While these changes should be a welcome update to most of our users, for those using different data types, you’re not out in the cold. Sentieon has abundant resources for building secondary analysis pipelines for all sorts of genomic data. In particular, Sentieon’s GitHub hosts an invaluable example scripts repository with example pipelines using Sentieon’s alignment and variant-calling algorithms. These scripts encompass a comprehensive list of data inputs, as well as useful scripts for benchmarking and post-processing. Additionally, users can find machine-learning model files for Illumina, MGI and Complete Genomics, Element Biosciences, Ultima Genomics, PacBio (with whom we’re delighted to have recently announced an exciting partnership), and Oxford Nanopore Technology. With these tools, users have an unprecedented number of options for inputs to VarSeq.
As always, we’re excited to hear your feedback and help you take advantage of our tools. Don’t hesitate to reach out to [email protected] with any questions!