
Last month, the researchers at Google DeepMind announced the release of AlphaMissense, a new missense prediction algorithm that leverages the protein structure prediction model AlphaFold to distinguish between benign and pathogenic missense variants (Cheng et al., 2023). AlphaFold is a model for the prediction of protein structures from amino acid sequences. During the development of AlphaMissense, the AlphaFold model was fine-tuned to identify variants commonly seen in human and primate populations. Variants predicted to be common are classified as benign, while variants predicted to be novel are classified as pathogenic. The authors report that AlphaMissense achieves 90% precision when used to classify a large subset of the ClinVar database.
While these results are impressive, we wanted to see for ourselves how this new algorithm stacks up against the other in-silico predictions that are currently available in VarSeq’s vast annotation library. We compared the predictions provided by AlphaMissese to 17 other missense prediction algorithms, using a subset of the ClinVar database as a benchmark dataset. In our experiments, AlphaMissense achieves an impressive balanced accuracy of 91.5% but is outperformed by BayesDel across all metrics.
Experimental Design
We compared AlphaMissense to the following in-silico prediction algorithms currently available in VarSeq:
- BayesDel (Feng, 2017)
- REVEL (Ioannidis et al., 2016)
- MetaSVM (Dong et al., 2015)
- MetaLR (Dong et al., 2015)
- MetaRNN (Li et al., 2022)
- DEOGEN2 (Raimondi et al., 2017)
- PROVEAN (Choi et al., 2012)
- FATHMM (Shihab et al., 2013)
- FATHMM-XF (Rogers et al., 2018)
- FATHMM-MKL (Shihab et al., 2015)
- PolyPhen-2 HVAR (Adzhubei et al., 2013)
- LRT (Chun et al., 2009)
- SIFT (Ng et al., 2003)
- LIST S2 (Malhis et al., 2020)
- PrimateAI (Sundaram et al., 2018)
- MutationTaster (Schwarz et al., 2010)
- CADD (Kircher et al., 2014)
For AlphaMissense, we classified variants as pathogenic if the score exceeded 0.564, and classified variants as benign if the score was below 0.34, as recommended by the authors. For CADD, variants were classified based on the raw scores using a threshold of 5 for a pathogenic classification and a threshold of 2 for a benign classification. For REVEL, we used a threshold of 0.5, which was the threshold that produced the best performance, according to Ioannidis et al. (2016). For all other prediction algorithms, we used the thresholds specified in the dbNSFP Function Predictions annotation source (Liu et al., 2020).
The performance of each algorithm was evaluated using the ClinVar repository for variant interpretations (Landrum et al., 2016). Variant classifications in ClinVar are contributed by both clinical and research laboratories, as well as expert panels comprised of medical professionals with a long-standing scope of work. Variants in ClinVar are classified using a five-tier system, and each variant is assigned a review status indicating the depth of evidence behind its interpretation, denoted by values ranging from 1 to 4 stars:
- The submission comes from a single submitter or multiple submitters with conflicting interpretations.
- Concurring interpretations are provided by at least two submitters with accompanying evidence.
- The interpretations have been reviewed by an expert panel.
- The ClinGen Steering Committee has evaluated and confirmed the evidence and assertion criteria, establishing its alignment with practice guidelines.
We restricted our analysis to high-quality ClinVar missense variants with a review status of at least 2 which had been classified as either Pathogenic (P), Likely Pathogenic (LP), Benign (B), or Likely Benign (LB). The truth set for ClinVar was defined as follows:
- Positives: Variants that are classified as Pathogenic (P) or Likely Pathogenic (LP)
- Negatives: Variants that are classified as Benign (B) or Likely Benign (LB)
Each classification provided by the prediction algorithms was categorized as either a True Positive, False Positive, True Negative, or False Negative as follows:
- True Positive (TP): Classified as P/LP in ClinVar and classified as P by the algorithm.
- False Positive (FP): Classified as B/LB in ClinVar but classified as P by the algorithm.
- True Negative (TN): Classified as B/LB in ClinVar and classified as B by the algorithm.
- False Negative (FN): Classified as P/LP in ClinVar but classified as B by the algorithm.
The number of variants falling into these categories was then used to compute the sensitivity, specificity, and balanced accuracy for each algorithm as follows:
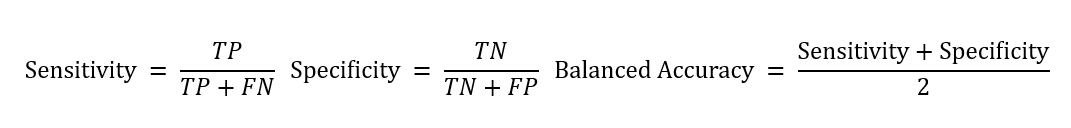
Results
Specific details about the ClinVar benchmark dataset are shown in the table below, including the total number of variants with each classification. This dataset was constructed using the ClinVar 2023-10-05 annotation track and contains a total of 35,468 variants.
| Total Variants in ClinVar 2023-08-03 | 2,254,932 |
| Variants with Classification of P/LP/B/LB | 1,070,385 |
| 2+ Star Variants with Required Classification | 191,149 |
| Missense Variants with Required Classification/Status | 35,468 |
| Pathogenic | 4,315 (12.2%) |
| Likely Pathogenic | 9,309 (26.2%) |
| Likely Benign | 14,017 (39.5%) |
| Benign | 7,827 (22.1%) |
The sensitivity, specificity, and balanced accuracy statistics for each algorithm are shown in Table 2, with all algorithms sorted by balanced accuracy.
| TP | FN | TN | FP | Sensitivity | Specificity | Balanced Accuracy | |
| BayesDel | 12,803 | 763 | 20,951 | 735 | 94.4% | 96.6% | 95.5% |
| AlphaMissense | 10,893 | 1,407 | 18,897 | 1,116 | 88.6% | 94.4% | 91.5% |
| REVEL | 12,632 | 1,047 | 19,573 | 2,162 | 92.3% | 90.1% | 91.2% |
| MetaSVM | 11,861 | 1,630 | 18,990 | 2,604 | 87.9% | 87.9% | 87.9% |
| MetaLR | 11,774 | 1,717 | 18,337 | 3,257 | 87.3% | 84.9% | 86.1% |
| PROVEAN | 12,051 | 1,672 | 17,092 | 4,719 | 87.8% | 78.4% | 83.1% |
| DEOGEN2 | 11,344 | 2,968 | 18,221 | 2,755 | 79.3% | 86.9% | 83.1% |
| FATHMM-XF | 11,660 | 749 | 14,270 | 5,994 | 94.0% | 70.4% | 82.2% |
| PolyPhen | 11,156 | 1,471 | 14,684 | 5,558 | 88.4% | 72.5% | 80.4% |
| MetaRNN | 13,053 | 502 | 14,017 | 7,827 | 96.3% | 64.2% | 80.2% |
| LRT | 11,253 | 1,152 | 12,521 | 6,009 | 90.7% | 67.6% | 79.1% |
| CADD | 348 | 260 | 11,732 | 13 | 57.2% | 99.9% | 78.6% |
| SIFT | 12,305 | 1,316 | 14,817 | 7,572 | 90.3% | 66.2% | 78.3% |
| MutationTaster | 13,253 | 371 | 12,433 | 9,411 | 97.3% | 56.9% | 77.1% |
| LIST S2 | 11,899 | 1,728 | 14,475 | 7,201 | 87.3% | 66.8% | 77.0% |
| FATHMM | 10,718 | 2,863 | 15,245 | 6,432 | 78.9% | 70.3% | 74.6% |
| FATHMM-MKL | 13,208 | 320 | 9489 | 12,188 | 97.6% | 43.8% | 70.7% |
| PrimateAI | 5,481 | 7,933 | 20,394 | 968 | 40.9% | 95.5% | 68.2% |
Using ClinVar as a benchmark, BayesDel achieves the highest balanced accuracy (95.5%), while performing extremely well in terms of both sensitivity and specificity, with all metrics exceeding 94%. AlphaMissense has the second-highest balanced accuracy at 91.5%, while also maintaining high sensitivity and specificity metrics of 88.6% and 94.4%, respectively. REVEL also performed well, achieving the third-highest balanced accuracy, with all metrics exceeding 90%. While AlphaMissense achieves slightly higher specificity and balanced accuracy than REVEL, the overall performance between these two algorithms is comparable.
Conclusion
In this blog post, we compared AlphaMissense to 17 other in-silico prediction algorithms, using ClinVar as a benchmark dataset. Of these algorithms, BayesDel, AlphaMissense, and REVEL demonstrated the highest overall performance, with all three algorithms achieving high balanced accuracy without sacrificing either sensitivity or specificity. These results are consistent with the findings of Tian et al., who also compared REVEL and BayesDel to a number of other prediction algorithms and found that these two algorithms outperformed competing methods in terms of both positive and negative predictive value (Tian et al., 2019).
While AlphaMissense demonstrated excellent performance, it was outperformed by BayesDel across all metrics. The superior performance of BayesDel is likely attributable to the algorithm’s incorporation of population frequency into its model, which allows it to more easily identify benign variants. The performance of AlphaMissense relative to BayesDel is impressive, given that it does not directly incorporate any population frequency information.
Golden Helix recently released a new annotation track containing the complete set of AlphaMissense predictions for all single nucleotide missense variants from 19,000 protein-coding genes for both GRCh37 and GRCh38 coordinates. These missense predictions make an excellent addition to VarSeq’s already extensive collection of functional prediction scores. I hope this blog post helped to give you an idea of how the AlphaMissense predictions compare to other in-silico prediction algorithms. If you have any questions about functional predictions in VarSeq, please don’t hesitate to reach out to us at [email protected].
References
- Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., … & Avsec, Ž. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science, eadg7492.
- Feng, B. J. (2017). PERCH: a unified framework for disease gene prioritization. Human mutation, 38(3), 243-251.
- Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., … & Sieh, W. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. The American Journal of Human Genetics, 99(4), 877-885.
- Dong, C., Wei, P., Jian, X., Gibbs, R., Boerwinkle, E., Wang, K., & Liu, X. (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human molecular genetics, 24(8), 2125-2137.
- Li, C., Zhi, D., Wang, K., & Liu, X. (2022). MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. Genome Medicine, 14(1), 115.
- Raimondi, D., Tanyalcin, I., Ferté, J., Gazzo, A., Orlando, G., Lenaerts, T., … & Vranken, W. (2017). DEOGEN2: prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic acids research, 45(W1), W201-W206.
- Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., & Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One, 7(10), e46688.
- Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., … & Gaunt, T. R. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Human mutation, 34(1), 57-65.
- Rogers, M. F., Shihab, H. A., Mort, M., Cooper, D. N., Gaunt, T. R., & Campbell, C. (2018). FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics, 34(3), 511-513.
- Shihab, H. A., Rogers, M. F., Gough, J., Mort, M., Cooper, D. N., Day, I. N., … & Campbell, C. (2015). An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics, 31(10), 1536-1543.
- Adzhubei, Ivan, Daniel M. Jordan, and Shamil R. Sunyaev. “Predicting functional effect of human missense mutations using PolyPhen‐2.” Current protocols in human genetics 76.1 (2013): 7-20.
- Chun, S., & Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome research, 19(9), 1553-1561.
- Ng, P. C., & Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research, 31(13), 3812-3814.
- Malhis, N., Jacobson, M., Jones, S. J., & Gsponer, J. (2020). LIST-S2: taxonomy based sorting of deleterious missense mutations across species. Nucleic acids research, 48(W1), W154-W161.
- Sundaram, L., Gao, H., Padigepati, S. R., McRae, J. F., Li, Y., Kosmicki, J. A., … & Farh, K. K. H. (2018). Predicting the clinical impact of human mutation with deep neural networks. Nature genetics, 50(8), 1161-1170.
- Schwarz, J. M., Rödelsperger, C., Schuelke, M., & Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods, 7(8), 575-576.
- Kircher, M., Witten, D. M., Jain, P., O’roak, B. J., Cooper, G. M., & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics, 46(3), 310-315.
- Liu, X., Li, C., Mou, C., Dong, Y., & Tu, Y. (2020). dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome medicine, 12(1), 1-8.
- Landrum, M. J., Lee, J. M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., … & Maglott, D. R. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research, 44(D1), D862-D868.
- Tian, Y., Pesaran, T., Chamberlin, A., Fenwick, R. B., Li, S., Gau, C. L., … & Qian, D. (2019). REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Scientific Reports, 9(1), 12752.



