Large-scale next-generation sequencing studies are becoming increasingly popular clinical and research tools. One enduring challenge for interpretion of these large amounts of data has been predicting the functional impact of genetic variants. Access to efficient computational tools for predicting the functional impact of variants is crucial to prioritizing the most potentially relevant variants in a dataset in a time-efficient manner. Some functional prediction tools take all kinds of variants into consideration, and others are more narrow in scope, focusing on variants that affect protein coding. VarSeq hosts a variety of functional prediction databases on our annotation server. The purpose of this article is to highlight the tools that are at your disposal for predicting the functional impact of single nucleotide variants (SNVs) insertions and deletions, to give a brief overview of their usage, and to show where to access them in the software.
Let’s start by looking at one of our most accessible tools for identifying gene impact, the RefSeq database. This database provides a summary of the computed interactions between a variant and its overlapping transcripts. Once a user annotates with RefSeq, they can identify loss of function and missense variants by pulling the Effect (Combined) field (Figure 1a) into the filter chain.
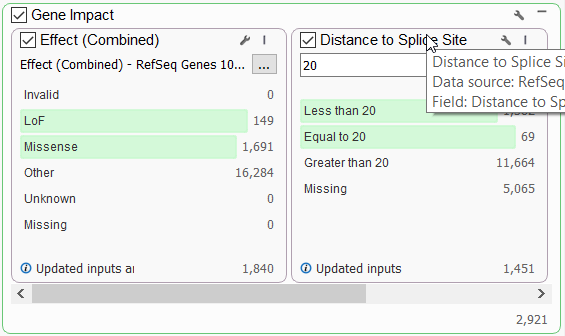
Variants that affect mRNA splicing could potentially impact protein product, and so will also be important to capture alongside the LOF and missense variants. These can also be generally accounted for by leveraging the Distance to Splice Site field (Figure 1b). Alternatively, to capture non-coding variants at a high level, a user could invert the Effect (combined) filter. After setting up this filter to follow either avenue, additional functional impact resources can be leveraged to be more granular in the type of variant that is being sought after.
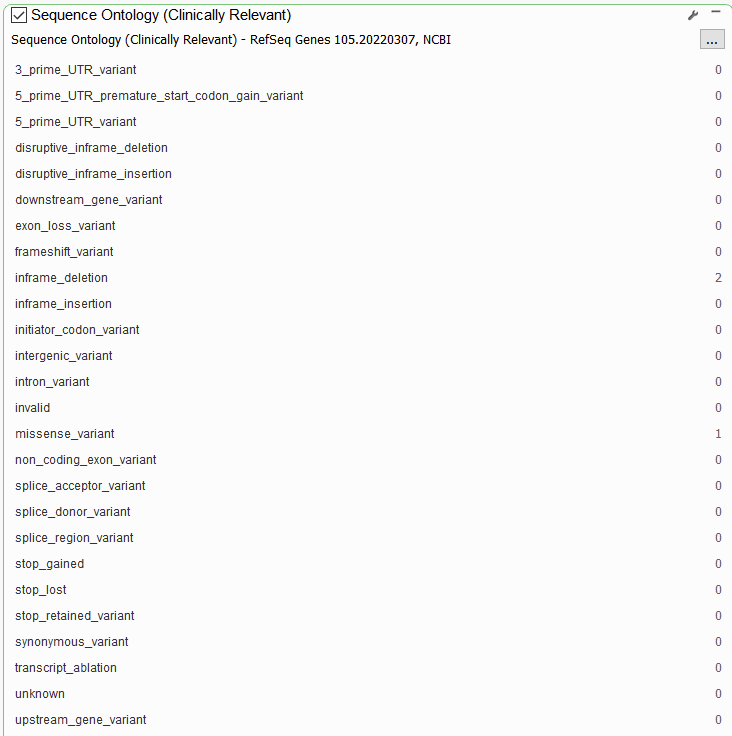
RefSeq also hosts more specific functional prediction algorithms for assessing the impact of a variant on the protein product and on splicing. The Sequence Ontology filter can be used to take a more granular look at functional impact than the Effect (Combined) filter (Figure 2).
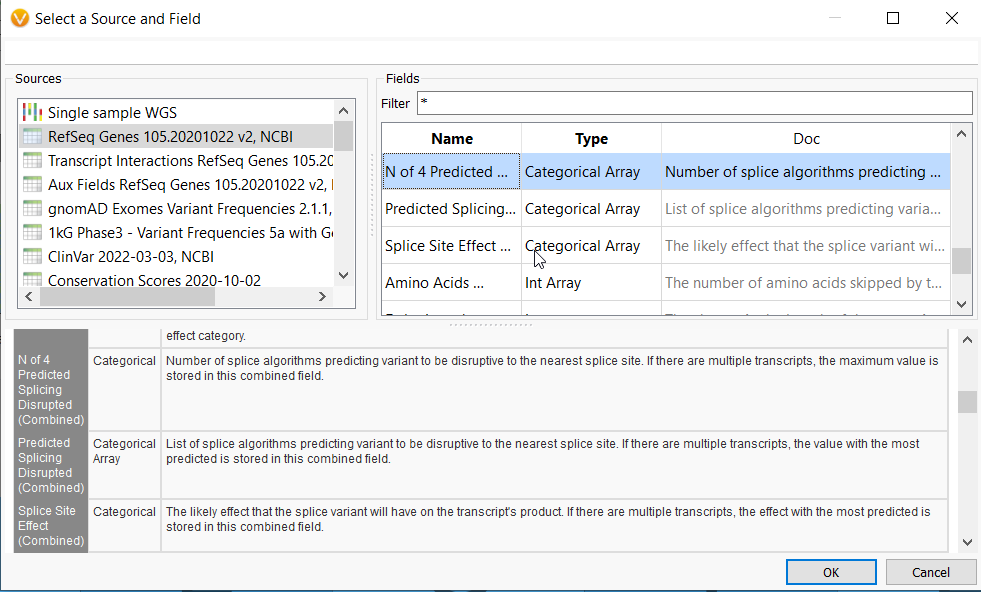
The N of 4 Predicted Splicing Disrupted field from RefSeq will predict how many of four top splice prediction tools indicate a variant will affect splicing, while the Predicted Splicing Disrupted allows you to choose the specific predictive algorithm to focus on, i.e., GeneSplicer, MaxEntScan, NNSplice, or PWM. The Splice Site Effect offers categories for the kind of splice site effect a variant may have, for example, donor loss or exon skipping, whether frame-shift or in-frame.
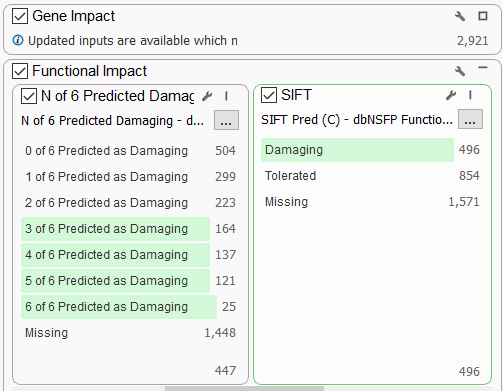
The dbSNP Functional Prediction Voting database is also very useful for identifying variants’ functional impact based on pooled evidence from a number of tools (Figure 4a). This allows you to identify damaging or tolerated predictions from Sift, Polyphen2, and MutationAssessor (only non-synonymous variants) and MutationTaster, FATHMM, and FATHMM MKL Coding, which cover a wider scope.
Wanting to become a VarSeq wizard?
A user can leverage the combined information using the N of 6 Predicted Damaging (or Tolerated) field as a filter. A conservative filter may be built by electing to keep variants with 3 or more of these algorithms predicting a damaging effect (Figure 4b).
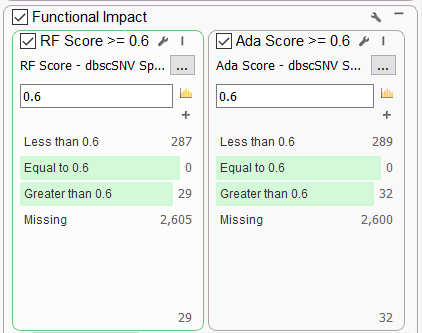
One very commonly requested set of functional predictions comes from the dbscSNV Splice Altering Predictions annotation maintained by Ensembl. This database contains pre-computed prediction scores for all SNVs in splice consensus regions. Users can leverage the Ada Score (based on ada-boost) and the RF Score (random forest), both ranging from 0 to 1, with a larger score indicating a higher probability that the variant will affect splicing.
Let’s end with two favorites that are often used together in a filtering strategy in VarSeq, especially when looking at whole genomes (Figure 6). The Rare Exome Variant Ensemble Learner (REVEL) is useful for predicting the functional impact and pathogenicity of missense variants. The REVEL score is based on a combination of scores from 13 separate tools, some of which were mentioned before: MutPred, FATHMM v2.3, VEST 3.0, PolyPhen-2, SIFT, PROVEAN, MutationAssessor, MutationTaster, LRT, GERP++, SiPhy, phyloP, and phastCons. Like the Ada and RF scores, The REVEL score ranges from 0 to 1, and the higher the score the greater the probability that the variant is pathogenic.
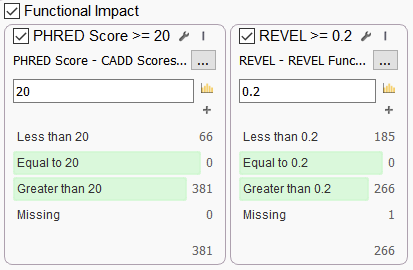
Combined Annotation Dependent Depletion (CADD) is one of the most widely known tools for predicting the functional impact of genetic variants. CADD offers Phred scale scores of the deleteriousness of human single nucleotide variants as well as insertion/deletions. CADD is quite broad in scope, integrating multiple annotations into one metric that addresses both coding and non-coding variants across a wide range of functional categories (Figure 6).
VarSeq provides both high-level and very granular approaches for predicting the functional impact of your genetic variants. As always, we can bring in certain custom annotations as well. I hope this article has helped to highlight the functional prediction capabilities of our VarSeq software. Please reach out to [email protected] for any additional questions or training on this topic.
Want to find out how VarSeq can fit into your variant analysis?