While VarSeq has always had excellent support for variant interpretation and analysis, we continue to find new edge cases in the clinical literature that improve our interpretation capabilities. In this blog, we will be covering some of the new improvements in VarSeq to support the interpretation of non-coding and splice site variants.
Transcript Annotation Improvements
Let’s start by covering some of the improvements to VarSeq’s transcript annotation algorithm. While VarSeq has always had the ability to annotate non-coding RNAs, by default, the transcript annotation algorithm would restrict its analysis to mRNAs, as the majority of non-coding transcripts are clinically uninteresting. However, a growing body of clinical literature describes pathogenic non-coding variants, including variants in promoter regions that don’t overlap any genes at all. As a result, we have updated our transcript annotation algorithm to better support the growing need to classify and interpret these variants. Our annotate transcripts algorithm now includes both non-coding RNAs and microRNAs by default, as well as variants 1,000 base pairs upstream or downstream from the nearest transcript. While this will be the default behavior for new templates, it should be noted that users must opt-in to the new settings when upgrading existing templates.
Splice Site Improvements
Another important update to the annotate transcript algorithm is the inclusion of novel splice predictions by default. While this does slow down annotation time by about 10%, these splice annotations have become an integral part of our clinical interpretation workflows and should be included for most use cases. We have also made several improvements to the splice prediction algorithms for this release. The most significant of these improvements are VarSeq’s new splice effect predictions, which provide information about the likely impact of the splice mutation on protein function.
We have also made important updates to how we recommend the ACMG criteria based on these splice predictions. Most importantly, we have moved our in-silico splice prediction recommendations to the PP3 criterion instead of the modified PVS1 criterion. Variants predicted to disrupt an existing splice site or introduce a novel splice site will now result in a recommendation of either PP3_Moderate or PP3_Strong. This change was made after a discussion with one of the authors of the updated PVS1 guidelines, as we are always striving to follow current best practices in our clinical workflows. Now that we have gone over these important changes let’s look at a couple of example variants in VSClinical.
Example 1 (RMRP n.-19_-3dup)
In this example, we have a 17 base pair duplication that is downstream from the gene RMRP.
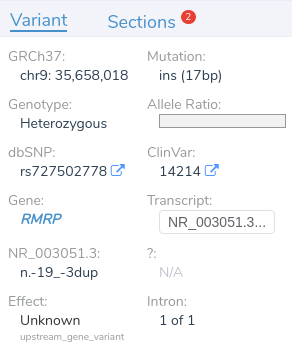
While two nearby genes could potentially be relevant here, VSClinical has picked RMRP, as it is the most clinically interesting of the two. This gene was chosen because it contains numerous previously classified ClinVar variants, including over 400 assessments. When multiple genes could potentially be relevant, VSClinical uses a set of advanced heuristics to select the most clinically relevant gene. However, users can always switch the interpretation to a different gene’s scope if the default selection is not the most clinically relevant for the current interpretation.
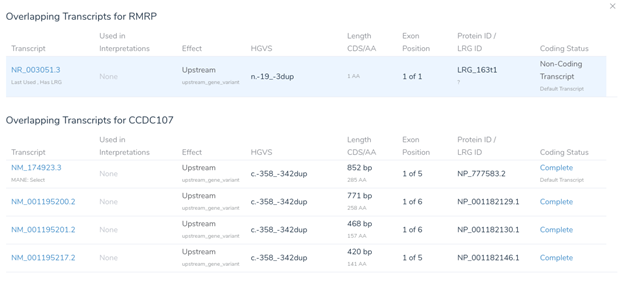
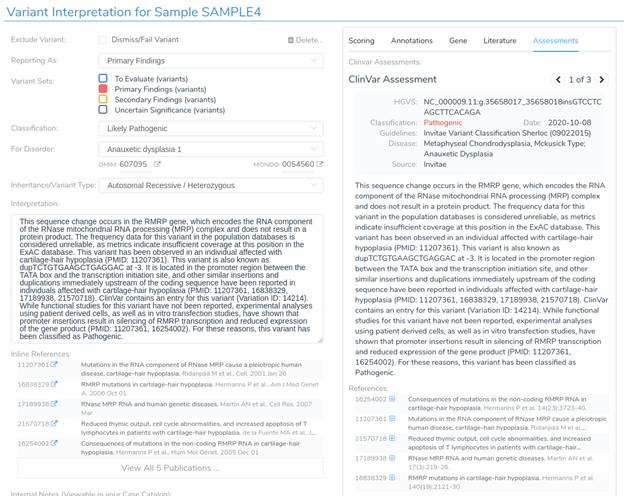
Looking at the recommended criteria, we can see that PM2 was recommended due to the variant’s low population frequency in GnomAD and 1kG. Additionally, PP5 has also been recommended, as the variant has been previously classified as pathogenic in ClinVar.

In the interpretation section, we can see that this variant has already been interpreted by three other labs, including Invitae and LabCorp. We can now examine these assessments and add the relevant information to our own interpretation, including all relevant publications provided by the assessment authors.
Example 2 (NF1 c.1261-19G>A)
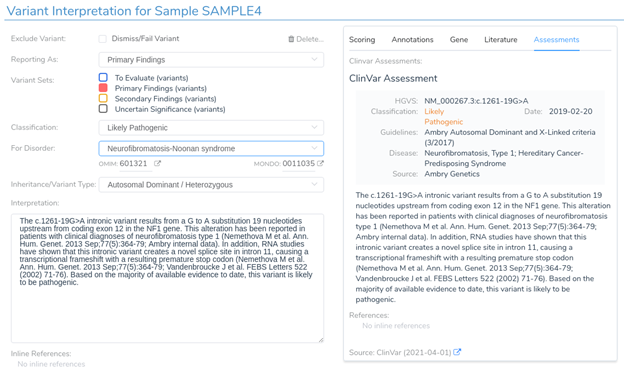
Next, we will look at an intronic substitution in the gene NF1. Normally, a variant like this would be ignored as it is 19 bases from the nearest exon boundary. However, in this case, the variant is predicted to introduce a novel acceptor splice site by 3 of the 4 splice prediction algorithms. It has been previously classified as pathogenic in ClinVar.

If we examine the PP3 section of VSClinical, we can see why we are providing these recommendations. The variant is predicted to cause a frameshift mutation, and 3 of the 4 splice prediction algorithms predict the introduction of a novel splice site. Notice that we present the number of algorithms that changed to predict splicing rather than the number of algorithms that simply predict splicing on the alternate sequence alone. This is an important distinction as we could have a position that was already predicted to be a splice site in the reference sequence.

Our splice site analysis view really helps to illustrate what is going on here. The blue boxes in the plot below indicate the presence of a novel splice site. We can see why a novel splice site is predicted here by comparing each algorithm’s score between the reference and alternate sequence.

If we return to the variant interpretation section, we can see that a previous clinical assessment has been provided for this variant in ClinVar. This assessment states that “RNA studies have shown that this intronic variant creates a novel splice site in intron 11, causing a transcriptional frameshift.” This further reinforces the predictions provided by our in-silico splice site algorithms and indicates that this variant is likely to introduce a novel splice site, resulting in a frameshift mutation.
Conclusion
I hope you enjoyed this discussion of the new improvements in VarSeq to support non-coding and splice site variants’ interpretation. If you have any questions about these new features, feel free to contact our technical support team at [email protected]. If you enjoyed this content, please check out some of our other blog posts, which contain important information and updates on our clinical interpretation capabilities.