Our latest VarSeq release is one of the largest we’ve ever had, boasting an extensive list of new features and improvements. As part of this release, we have dramatically expanded our support for splice site analysis. This includes improvements to our novel splice site algorithm and support for splice site effect prediction along with several other small improvements.
Novel Splice Site Prediction
We have made several improvements to our novel splice site prediction annotation. Previously, the novel splice site annotation would report any increase in the number of algorithms predicting splicing as a novel splice site. This resulted in many existing splice sites being predicted as novel due to nearby mutations altering the splice motif. The novel splice site algorithm has now been updated to no longer consider changes to existing splice sites.
We have also added a new field to our novel splice site annotation which reports the number of algorithms that have changed their prediction due to a given mutation. This field is extremely useful when assessing the reliability of a given novel splice prediction. For instance, a potential novel splice site for which only a single algorithm changed its prediction would be more likely to be a false positive when compared to a novel splice site in which all algorithms changed to predict splicing.

Splice Effect Predictions
The most significant addition to our splice site analysis capabilities is our splice site effect predictions. When running transcript annotations, each splice disrupting variant is classified into one of three categories based on it’s predicted effect on the gene product:
- Donor Loss
- Exon Skipping Frameshift
- Exon Skipping Inframe
While variants that disrupt a nearby donor splice site are simply classified as “Donor Loss”, variants that disrupt an acceptor site are classified as either frameshift or inframe based on the affected exon. These predicted effects are used to inform the recommended criteria in the ACMG workflow within VSClinical. The following examples illustrate the distinction between these different splice effects and show how these different effects alter the recommendations provided by VSClinical.

The figure below shows the donor loss variant NM_006015.6:c.1920+5G>A.
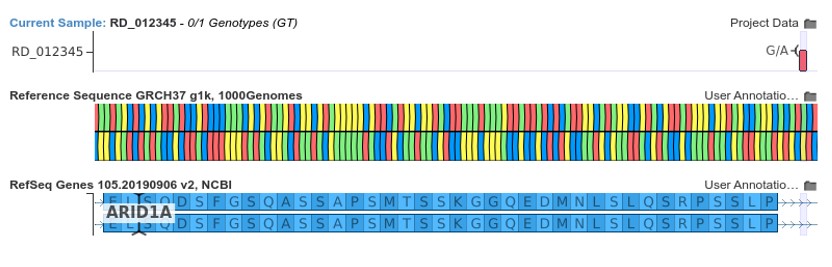
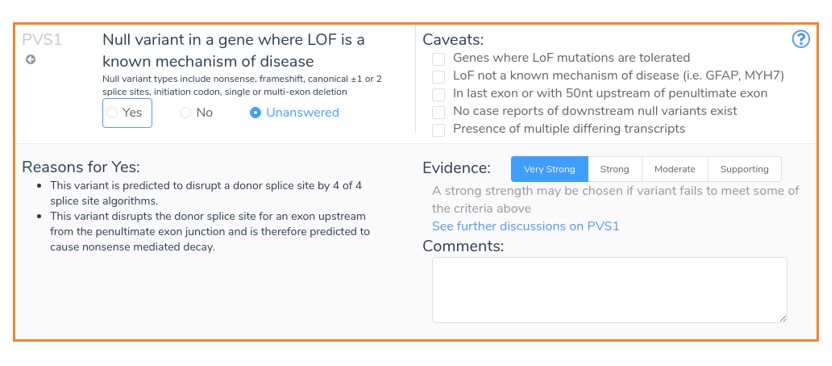
This mutation results in the loss of the donor splice site of exon 4 in the gene ARID1A. In general, donor loss variants that occur upstream from the penultimate exon junction are assumed to cause nonsense-mediated decay. Thus, VSClinical recommends the application of PVS1 for such variants as shown below, provided that all splice site prediction algorithms agree that the donor site is disrupted.
For our second example, we will look at the exon skipping frameshift mutation NM_153006.3:c.916-3C>G.

This mutation results in the loss of the acceptor splice site preceding exon 4 in the gene NAGS. Loss of this exon will result in a frameshift and is predicted to cause nonsense-mediated decay, as it is upstream from the last coding exon of the gene. Because all splice site prediction algorithms agree that this splice site is disrupted, VSClinical recommends PVS1.

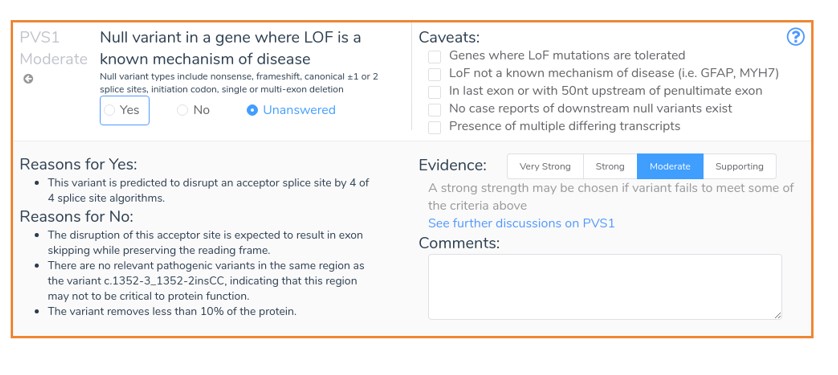
In our final example, we will look at the inframe exon skipping mutation NM_001077350.3:c.1352-3_1352-2insCC.

In this example, an intronic insertion results in the loss of an acceptor site upstream from exon 13 of the reverse-strand NPRL3 gene. This mutation is expected to result in inframe exon skipping and is therefore not predicted to cause nonsense-mediated decay. Since this variant is expected to remove less than 10% of the protein and there are no previously classified pathogenic variants in this region, VSClinical recommends PVS1_Moderate. This recommendation is based on the updated PVS1 guidelines, which were published in 2018. With the new release of VarSeq, these updated guidelines are fully supported by VSClinical, and we hope to fully explore these updates in a future blog post.
Other Improvements
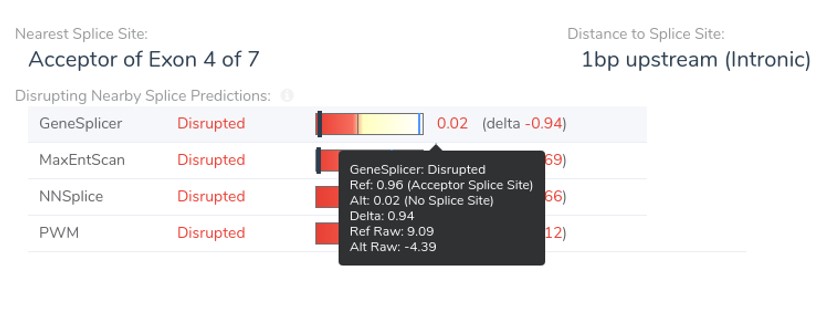
In addition to the updates discussed above, we have made two other minor improvements to our splice site algorithms. First, the “Distance to Splice Site” field has been updated to account for all combinations of a variant being before or after the splice site, on either the forward or reverse strands. This distance calculation is now the number of bases that would be required to move the variant to overlap the canonical splice site. This is illustrated by the figure below, which shows the donor loss variant NM_006015.6:c.1920+5G>A discussed above along with its distance to the clinically relevant splice site in the variant’s table.

Second, we have added the raw scores for the MaxEntScan and GeneSplicer prediction algorithms to the VSClinical splice site display. Previously, we only displayed normalized scores which had been adjusted to be between 0 and 1. Because the NNSplice and PWM algorithms already produce raw scores between 0 and 1, no distinction exists between raw and normalized values for these algorithms. The raw Genesplicer scores for the frameshift splice acceptor variant NM_153006.3:c.916-3C>G, are shown in the figure below.
Conclusion
If you want to know more about our splice site analysis capabilities or any other features in VarSeq, please contact us at [email protected]. We have also released a detailed breakdown of our recent release notes on VarSeq version 2.2.2 on our website. Feel free to also check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.