In a recent webcast, users were exposed to some new features upcoming in the next release of VarSeq. In this update, we will use an example de Novo CNV in a cardiomyopathy panel in VarSeq. There is a long list of new tools and polishes to the software but one major upgrade is the inclusion of the ACMG and ClinGen joint consensus guidelines for processing copy-number variants. Not only did the webcast cover fundamentals of the guidelines, but also demonstrated the user experience with some direct examples. One example was for a de novo heterozygous deletion in DSG2. This particular patient has suffered heart complications from a young age and was eventually diagnosed with arrhythmogenic right ventricular cardiomyopathy via ECG and MRI imaging. The goal was then to isolate the underlying genetic factor which is where VarSeq comes in.
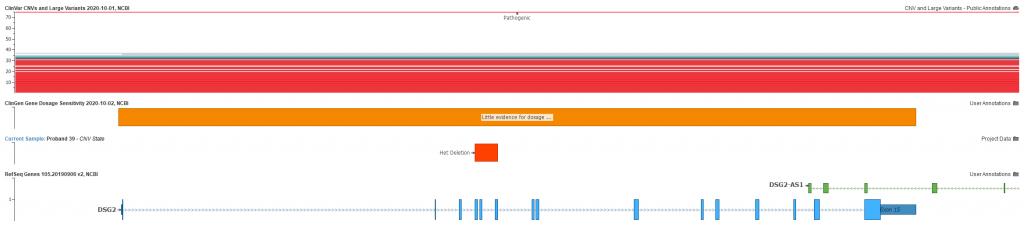
The sample was collected and run through a whole exome analysis which included the BAM and VCF files imported into VarSeq. No obvious contributing SNPs or indels came through the tertiary filtering, however this single heterozygous deletion was present. Figure 1 shows the plotted CNV overlapping exons 4-6 of the DSG2 gene and known pathogenic CNVs present in ClinVar. Notice also the ClinGen record for little evidence of dosage pathogenicity for DSG2. This evidence greatly influences the final classification of the CNV following the aforementioned guidelines.
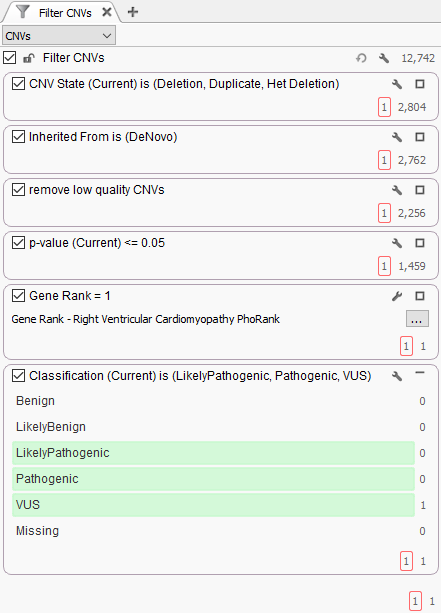
The filtering strategy to find this event can be seen in Figure 2. The first step was to focus on events detected in the proband, detect and keep de novo CNVs, ensure the calls are high quality and eliminate false positives, plus focus the search for events in genes with a strong association to the targeted phenotype. Additionally, the new ACMG CNV classification algorithm was implemented in the filter and this isolated het deletion was labeled as a variant of uncertain significance (VUS).
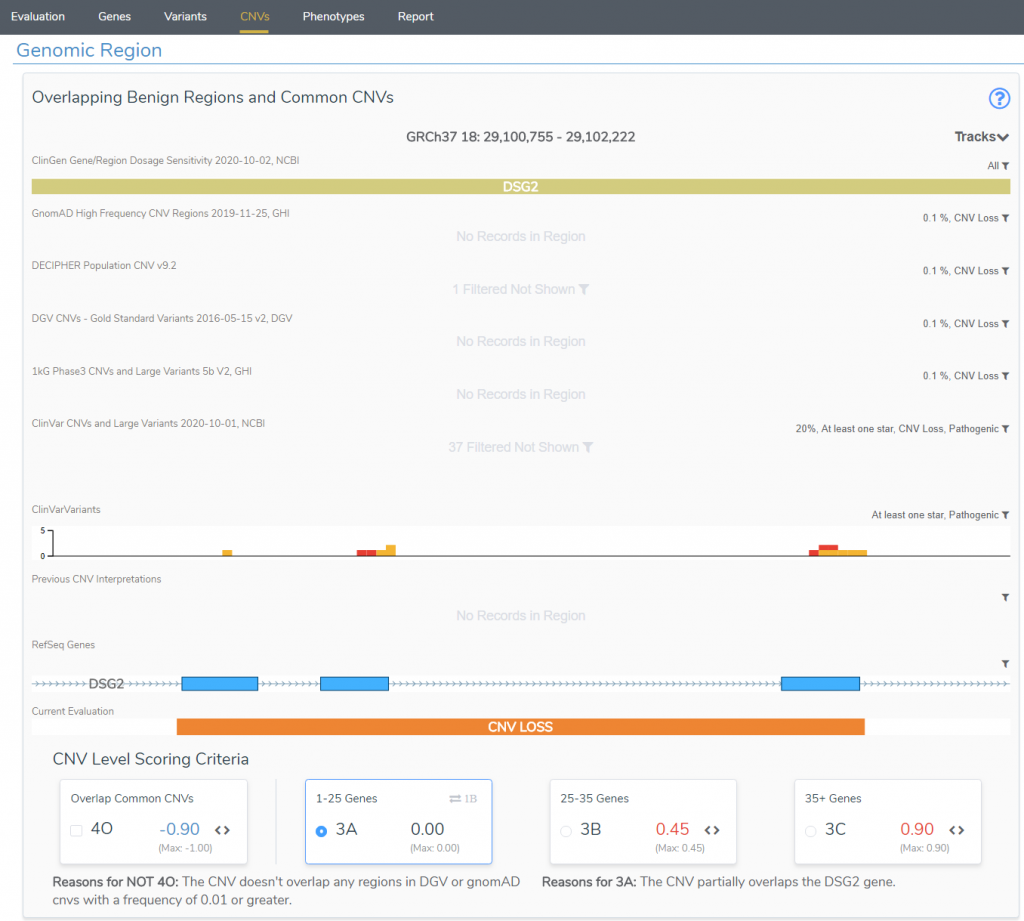
As previously mentioned, known dosage sensitivity for a gene in ClinGen contributes greatly to a CNVs classification. However, if evidence is absent or weak for a gene, users will need to start building records of sample cases with overlapping events to establish the dosage sensitivity themselves. VarSeq also seeks to simplify this process. Within VarSeq is the clinical interpretation hub, VSClinical, which will assist the user with processing all relevant criteria for any variant while directly following the appropriate guidelines. Figure 3 shows the first presentation of these relevant criteria. This plot is a quick reference to answer some simple questions such as, is this variant common? In this case no, this het deletion in DSG2 has no overlapping CNVs with tracks like DGV or GnomAD, for example. This is also the reason why you will see the criteria 4O unselected. However, 3A is being applied in this case, since the event spans a few exons of a single gene rather than an increased span of 25-35 genes (3B +0.45 score), or 35+ genes (3C +0.9 score). This plot also shows known pathogenic variants/CNVs seen in ClinVar, and this is going to be relevant to the next criteria review.
Being that there is little evidence for dosage sensitivity in ClinGen, section 2 of the guidelines can be skipped. Instead, we will focus on sections 4 and 5 which connect the dots of any gene variants and the relevant disorder. Section 4 will allow users to submit any observed/documented variants relevant for the gene. This includes not only the samples processed in VarSeq but also any literature with documented cases. This is crucial in that if dosage sensitivity is not already known, users can start compiling the final score across multiple samples or records and in turn build their own dosage sensitivity conclusions. Figure 4 shows the section 4 wizard for scoring criteria of this de novo event.

The first step in the path is stating that inheritance is known, which is a confirmed de novo event, and in a gene consistent with the phenotype. The final result is that this variant is currently scored with 0.30 as uncertain significance. However, if this event or similar events in the same gene start to accumulate in future samples, these records will add up and potentially change the classifications over time. For example, if two other samples are seen for a similar event, each adding 0.3 scores, the final tally would reach 0.9, or likely pathogenic.

Of course, the crucial factor here is to record these events in your VarSeq assessment catalog that also records each unique sample. Golden Helix seeks to simplify your navigation through this sample and variant collection and evaluation not only with tools like VSClinical, but also optimize the review of this content with our genomic repository tool VSWarehouse. The goal of this blog was to highlight some of these new powerful tools in our upcoming release, but please reach out to us directly at [email protected] if you would like to test the software yourself or see a more comprehensive demonstration.