As I mentioned in the first part of this series, Sentieon allows users to call somatic variants against a matched normal sample and a tumor-only analysis.
Utilization of a Tumor-Normal Workflow
In addition to the fundamental process of alignment and variant calling, there are a few more steps that will improve the quality of your secondary analysis. Figure 1 (below) is an excellent overview of the process.
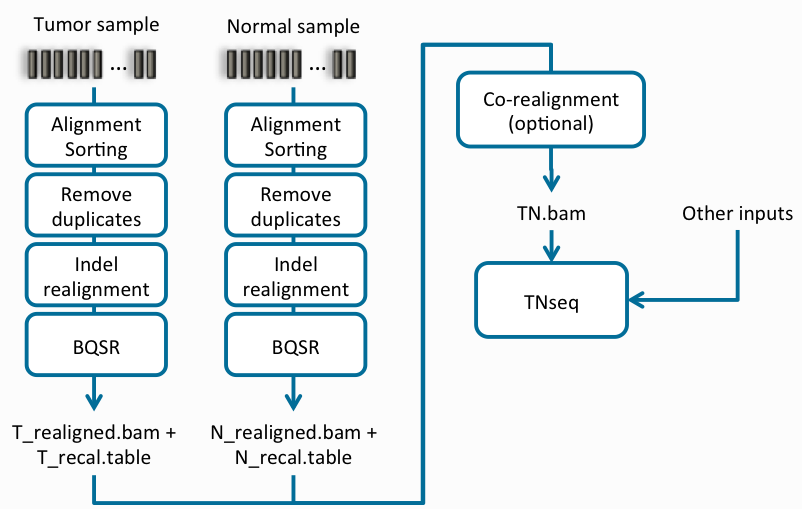
The first step in this process is to align all reads in the FASTQ file against the reference sequence (Alignment Sorting in Figure 1).
Next is the optional step of removing duplicated reads resulting from PCR amplification. For users running amplicon data, it is recommended to skip this step since most reads aligned will be PCR amplified.
Following deduplication of reads are the quality control steps to improve alignment; indel realignment and base quality score recalibration. The process of indel realignment leverages a VCF file (Mills and 1000G gold standard indels) containing known indel variants and users can also annotate against dbSNP and known somatic variants present in COSMIC. These annotations come shipped with the Sentieon package. As you can see in Figure 1 (above), this process is run independently for both the normal and tumor samples.
The final stage of the TNseq workflow undergoes an indel co-realignment of the tumor + normal sample as an optional step (can run as separate samples), and the last step is to call the variants. The output of this process is a VCF of potential somatic variants that remain after the exclusion of germline variants present in the normal sample.
We hope you found this to be a helpful, simple overview of how to process tumor-normal variant calling with Sentieon, which is supported for both the GRCh37 and 38 assemblies. Next, we show how to call somatic variants using a tumor sample with no matching normal – take a look!